Multiplexed assays of variant effect analysis with `mutscan`
Charlotte Soneson and Michael Stadler
28 April 2026
Source:vignettes/mutscan.Rmd
mutscan.RmdIntroduction
mutscan provides functionality for processing and
visualizing Multiplexed Assays of Variant Effect (MAVE) and other
similar types of data, starting from single-end or paired-end FASTQ
files. A broad range of library designs can be processed, encompassing
experiments considering either a single protein or combinations of
multiple proteins (e.g., aimed at studying protein-protein
interactions).
The figure below provides a high-level overview of the
mutscan functionality, which will be described in more
detail in the following sections.
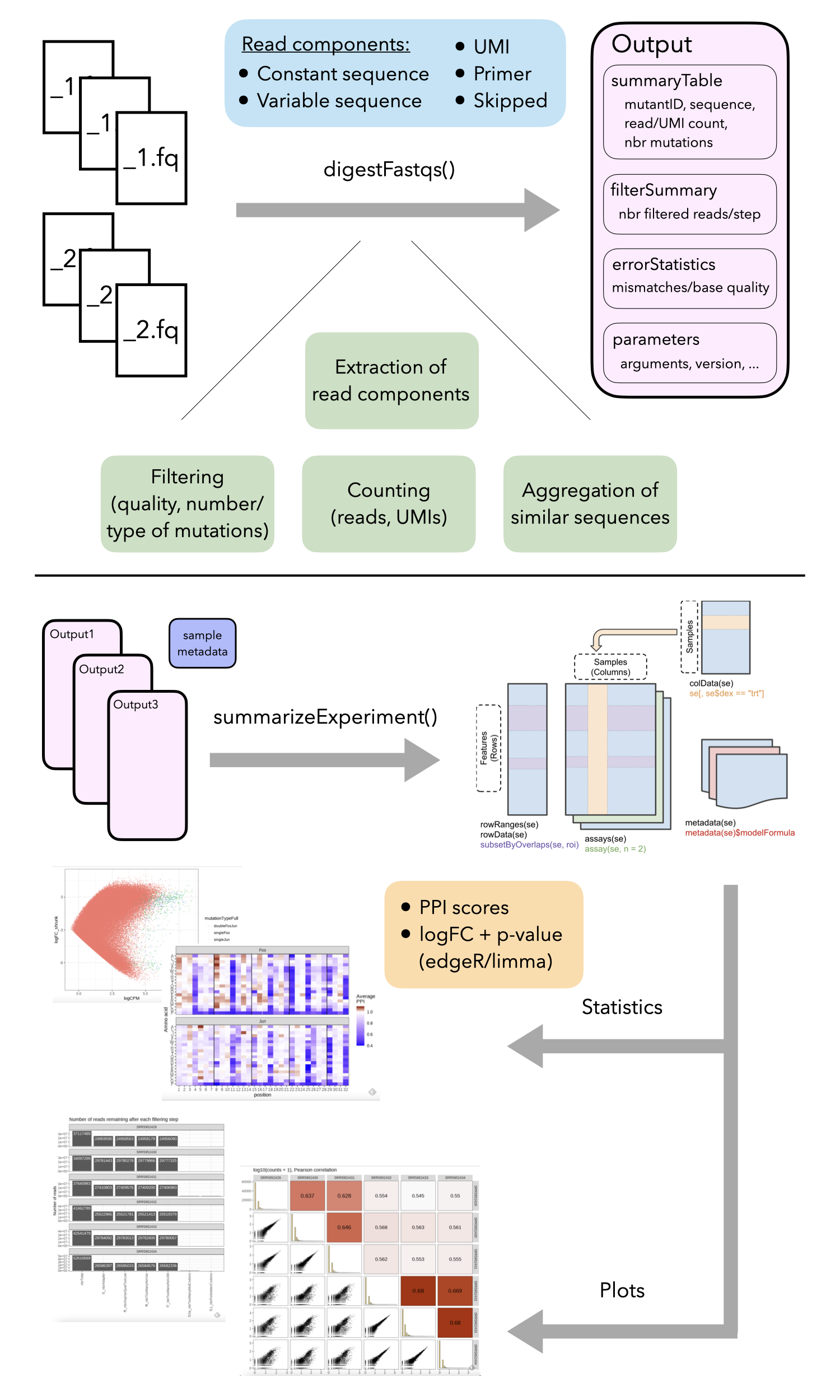
Overview of the mutscan functionality. The
digestFastqs() function processes each sequencing library,
possibly consisting of multiple (pairs of) FASTQ files, separately, and
generates an output object containing, among other things, the count
table and a summary of the filtering steps. The
summarizeExperiment() function takes one or more of these
objects and combines them into a SummarizedExperiment
object, that can then be used for downstream analysis such as plotting
and statistical testing.
Example data
The mutscan package contains several small example FASTQ
files representing different types of experiments:
datadir <- system.file("extdata", package = "mutscan")
dir(datadir)
#> [1] "cisInput_1.fastq.gz" "cisInput_2.fastq.gz"
#> [3] "cisOutput_1.fastq.gz" "cisOutput_2.fastq.gz"
#> [5] "GSE102901_cis_se.rds" "leujunt0_1.fastq.gz"
#> [7] "leujunt0_2.fastq.gz" "multipleVariableRegions_R1.fastq.gz"
#> [9] "multipleVariableRegions_R2.fastq.gz" "multipleVariableRegions_truth.rds"
#> [11] "transInput_1.fastq.gz" "transInput_2.fastq.gz"
#> [13] "transOutput_1.fastq.gz" "transOutput_2.fastq.gz"-
transInput_{1,2}.fastq.gz,transOutput_{1,2}.fastq.gz- data from TRANS experiment in [@Diss2018]. The forward and reverse reads correspond to the mutated FOS and JUN sequences, respectively. Each read consists of a UMI sequence, followed by a constant sequence and the variable region. -
cisInput_{1,2}.fastq.gz,cisOutput_{1,2}.fastq.gz- data from CIS experiment in [@Diss2018]. The forward and reverse reads both correspond to the mutated FOS sequence. Each read consists of a UMI sequence, followed by a constant sequence and the variable region. -
GSE102901_cis_se.rds- aSummarizedExperimentobject obtained by processing the full CIS data from [@Diss2018]. -
leujunt0_{1,2}.fastq.gz- unpublished data; the forward read corresponds to an unmutated sequence of one of 46 leucine zipper sequences, and the reverse read corresponds to mutated JUN sequences. Each read contains a (frame-shifted) primer sequence, followed by the variable region.
Processing Multiplexed Assays of Variant Effect data
Read composition specification
The main function for processing the Multiplexed Assays of Variant
Effect FASTQ files is digestFastqs(). This function
requires the specification of paths to compressed FASTQ file(s) and the
composition of the reads in these files. The composition is specified by
the user, and is given in the form of a character string indicating the
parts constituting the respective read, and an integer vector specifying
the lengths of the individual parts. The permitted components are:
-
S(skip) - nucleotide(s) to skip -
U(UMI) - UMI sequence -
C(constant) - constant sequence, can be used e.g. to estimate the sequencing error rate if available -
V(variable) - nucleotides corresponding to the variable region -
P(primer) - a primer sequence, which may appear in a nonspecified position in the read but whose position defines the start or end position of other components.
A read can have several components of the same type (e.g., skipped
nucleotides both in the beginning or the end, or two variable regions
separated by a primer). In such cases, mutscan will
concatenate all components of the same type in the processing, while
recording the lengths of the individual components.
As an example, a read with the following composition:
[1 skipped nt] - [10 nt UMI] - [18 nt constant sequence] - [96 nt variable region]
would be specified to digestFastqs() by an elements
string "SUCV", and an element length vector
c(1, 10, 18, 96).
The package can also accommodate designs with primer sequences. In this situation, the primer acts as an ‘anchor’, and the read composition before and after the primer is specified. For example, a read with the following composition:
[unknown sequence] - [10 nt primer] - [variable region, constituting the remainder of the read]
would be represented by an elements string "SPV", and an
element length vector c(-1, 10, -1), where the -1 indicates
that the corresponding read part consists of the remaining part of the
read, not accounted for by any of the other specified parts. In
addition, the sequence of the primer must be specified, and any read
where the primer is not present will be discarded.
The forward and reverse reads can have different compositions. The user can also specify whether the variable parts of the forward and/or reverse reads should be reverse complemented before being processed, and whether the variable regions of the forward and reverse reads should be merged into a single consensus sequence.
Filtering
In addition to reading the FASTQ files, the
digestFastqs() function will perform a series of filtering
steps, in the following order:
- Search for perfect matches to forward/reverse adapter sequences, filter out the read pair if a match is found in either the forward or reverse read.
- Search for perfect matches to forward/reverse primer sequences, filter out the read pair if not both are found.
- Filter out reads whose length is not compatible with the indicated composition.
- If forward and reverse variable regions should be merged, filter out read pairs where no valid overlap could be found.
- Filter out read pair if the average quality in the variable region
is below
avePhredMinin either the forward or reverse read (or the consensus sequence if they are merged). - Filter out read pair if the number of Ns in the variable region
exceeds
variableNMax. - Filter out read pair if the number of Ns in the combined forward and
reverse UMI sequence exceeds
umiNMax. - If wild type (reference) sequences are provided (via
wildTypeForwardand/orwildTypeReverse), filter out reads if there are multiple best matches among the provided wild type sequences. - Filter out read pair if any mutated base has a quality below
mutatedPhredMin. - Filter out read pair if the number of mutated codons exceeds
nbrMutatedCodonsMaxor the number of mutated nucleotides exceedsnbrMutatedBasesMax. - Filter out read pair if any of the mutated codons match any of the
codons encoded by
forbiddenMutatedCodons. - Filter out read pair if there are too many mutations in the constant sequence, or if there are multiple equally close best hits among the provided constant sequences.
Reads that are filtered out in one step will not be processed further (and thus will not contribute to the count of reads being filtered out for any other reasons).
Processing TRANS data
Here, we illustrate the processing of the provided TRANS experiment
example data. We filter out reads with any adapter match, an average
phred quality below 20, any Ns in the UMI or variable sequence, more
than one mutated codon, or a mutated codon ending with A or T
(represented by the NNW value of the
forbiddenMutatedCodonsForward and
forbiddenMutatedCodonsReverse arguments). In order to
identify the number of mutations, we have to provide the wild type
sequences of FOS and JUN (forward and reverse reads,
respectively). This will additionally name the final mutants according
to the positions and sequences of the mutated codons.
transInput <- digestFastqs(
fastqForward = file.path(datadir, "transInput_1.fastq.gz"),
fastqReverse = file.path(datadir, "transInput_2.fastq.gz"),
mergeForwardReverse = FALSE,
revComplForward = FALSE,
revComplReverse = FALSE,
adapterForward = "GGAAGAGCACACGTC",
adapterReverse = "GGAAGAGCGTCGTGT",
elementsForward = "SUCV",
elementsReverse = "SUCV",
elementLengthsForward = c(1, 10, 18, 96),
elementLengthsReverse = c(1, 8, 20, 96),
constantForward = "AACCGGAGGAGGGAGCTG",
constantReverse = "GAAAAAGGAAGCTGGAGAGA",
avePhredMinForward = 20,
avePhredMinReverse = 20,
variableNMaxForward = 0,
variableNMaxReverse = 0,
umiNMax = 0,
wildTypeForward = c(FOS = "ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA"),
wildTypeReverse = c(JUN = "ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT"),
nbrMutatedCodonsMaxForward = 1,
nbrMutatedCodonsMaxReverse = 1,
forbiddenMutatedCodonsForward = "NNW",
forbiddenMutatedCodonsReverse = "NNW",
mutNameDelimiter = ".",
constantMaxDistForward = -1,
constantMaxDistReverse = -1,
verbose = FALSE
)The digestFastqs() function returns a list with four
elements. The parameters list records all parameter values
used during the processing, as well as the mutscan version
and time of processing.
transInput$parameters
#> $fastqForward
#> [1] "/Users/runner/work/_temp/Library/mutscan/extdata/transInput_1.fastq.gz"
#>
#> $fastqReverse
#> [1] "/Users/runner/work/_temp/Library/mutscan/extdata/transInput_2.fastq.gz"
#>
#> $mergeForwardReverse
#> [1] FALSE
#>
#> $minOverlap
#> [1] 0
#>
#> $maxOverlap
#> [1] 0
#>
#> $minMergedLength
#> [1] 0
#>
#> $maxMergedLength
#> [1] 0
#>
#> $maxFracMismatchOverlap
#> [1] 1
#>
#> $greedyOverlap
#> [1] TRUE
#>
#> $revComplForward
#> [1] FALSE
#>
#> $revComplReverse
#> [1] FALSE
#>
#> $elementsForward
#> [1] "SUCV"
#>
#> $elementLengthsForward
#> [1] 1 10 18 96
#>
#> $elementsReverse
#> [1] "SUCV"
#>
#> $elementLengthsReverse
#> [1] 1 8 20 96
#>
#> $adapterForward
#> [1] "GGAAGAGCACACGTC"
#>
#> $adapterReverse
#> [1] "GGAAGAGCGTCGTGT"
#>
#> $primerForward
#> [1] ""
#>
#> $primerReverse
#> [1] ""
#>
#> $wildTypeForward
#> FOS
#> "ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA"
#>
#> $wildTypeReverse
#> JUN
#> "ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT"
#>
#> $constantForward
#> [1] "AACCGGAGGAGGGAGCTG"
#>
#> $constantReverse
#> [1] "GAAAAAGGAAGCTGGAGAGA"
#>
#> $avePhredMinForward
#> [1] 20
#>
#> $avePhredMinReverse
#> [1] 20
#>
#> $variableNMaxForward
#> [1] 0
#>
#> $variableNMaxReverse
#> [1] 0
#>
#> $umiNMax
#> [1] 0
#>
#> $nbrMutatedCodonsMaxForward
#> [1] 1
#>
#> $nbrMutatedCodonsMaxReverse
#> [1] 1
#>
#> $nbrMutatedBasesMaxForward
#> [1] -1
#>
#> $nbrMutatedBasesMaxReverse
#> [1] -1
#>
#> $forbiddenMutatedCodonsForward
#> [1] "AAA" "AAT" "ACA" "ACT" "AGA" "AGT" "ATA" "ATT" "CAA" "CAT" "CCA" "CCT"
#> [13] "CGA" "CGT" "CTA" "CTT" "GAA" "GAT" "GCA" "GCT" "GGA" "GGT" "GTA" "GTT"
#> [25] "TAA" "TAT" "TCA" "TCT" "TGA" "TGT" "TTA" "TTT"
#>
#> $forbiddenMutatedCodonsReverse
#> [1] "AAA" "AAT" "ACA" "ACT" "AGA" "AGT" "ATA" "ATT" "CAA" "CAT" "CCA" "CCT"
#> [13] "CGA" "CGT" "CTA" "CTT" "GAA" "GAT" "GCA" "GCT" "GGA" "GGT" "GTA" "GTT"
#> [25] "TAA" "TAT" "TCA" "TCT" "TGA" "TGT" "TTA" "TTT"
#>
#> $useTreeWTmatch
#> [1] FALSE
#>
#> $collapseToWTForward
#> [1] FALSE
#>
#> $collapseToWTReverse
#> [1] FALSE
#>
#> $mutatedPhredMinForward
#> [1] 0
#>
#> $mutatedPhredMinReverse
#> [1] 0
#>
#> $mutNameDelimiter
#> [1] "."
#>
#> $constantMaxDistForward
#> [1] -1
#>
#> $constantMaxDistReverse
#> [1] -1
#>
#> $umiCollapseMaxDist
#> [1] 0
#>
#> $filteredReadsFastqForward
#> [1] ""
#>
#> $filteredReadsFastqReverse
#> [1] ""
#>
#> $maxNReads
#> [1] -1
#>
#> $nThreads
#> [1] 1
#>
#> $chunkSize
#> [1] 100000
#>
#> $maxReadLength
#> [1] 1024
#>
#> $processingInfo
#> [1] "Processed by mutscan v1.3.0 on 2026-04-28 18:01:10.756292"The filterSummary data.frame contains a summary of the
number of reads filtered out in each step. Note that for any given
filtering step, only the reads retained by the previous filters are
considered. The numbers in the filter column names indicate the order of
the filters.
transInput$filterSummary
#> nbrTotal f1_nbrAdapter f2_nbrNoPrimer f3_nbrReadWrongLength
#> 1 1000 314 0 0
#> f4_nbrNoValidOverlap f5_nbrAvgVarQualTooLow f6_nbrTooManyNinVar
#> 1 0 7 0
#> f7_nbrTooManyNinUMI f8_nbrTooManyBestWTHits f9_nbrMutQualTooLow
#> 1 0 0 0
#> f10a_nbrTooManyMutCodons f10b_nbrTooManyMutBases f11_nbrForbiddenCodons
#> 1 392 0 8
#> f12_nbrTooManyMutConstant f13_nbrTooManyBestConstantHits nbrRetained
#> 1 0 0 279The summaryTable provides the number of reads and unique
UMIs observed for each variable sequence pair. The underscore in the
strings in the sequence column indicate the separation between the
forward and reverse wild type sequences. In addition, the table contains
a column mutantName, which provides a shorthand notation
for each mutant. The format of the values in this column is a
combination of WTS.xx.NNN (separated by _),
where WTS provides the name of the closest matching wild
type sequence (if only one, unnamed wild type sequence is provided, the
name will be f/r depending on if it corresponds to the
forward/reverse read, respectively). The xx part indicates
the mutated codon or nucleotide number, and NNN gives the
observed sequence for the mutated codon or nucleotide. A sequence
without mutations is named WTS.0.WT, where, again,
WTS is the name of the matching wild type sequence.
head(transInput$summaryTable)
#> mutantName
#> 1 FOS.0.WT_JUN.13.CCC
#> 2 FOS.0.WT_JUN.13.CTC
#> 3 FOS.0.WT_JUN.2.TCC
#> 4 FOS.0.WT_JUN.20.ACC
#> 5 FOS.0.WT_JUN.30.AGG
#> 6 FOS.0.WT_JUN.30.GGG
#> sequence
#> 1 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA_ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAACCCCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> 2 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA_ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAACTCCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> 3 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA_ATCTCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> 4 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA_ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGACCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> 5 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA_ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGAGGCAGCTT
#> 6 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA_ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGGGCAGCTT
#> nbrReads maxNbrReads nbrUmis nbrMutBases nbrMutCodons nbrMutAAs varLengths
#> 1 1 1 1 2 1 1 96_96
#> 2 1 1 1 3 1 1 96_96
#> 3 1 1 1 1 1 1 96_96
#> 4 1 1 1 1 1 1 96_96
#> 5 1 1 1 3 1 1 96_96
#> 6 1 1 1 2 1 1 96_96
#> mutantNameBase mutantNameCodon
#> 1 FOS.0.WT_JUN.37.C_JUN.39.C FOS.0.WT_JUN.13.CCC
#> 2 FOS.0.WT_JUN.37.C_JUN.38.T_JUN.39.C FOS.0.WT_JUN.13.CTC
#> 3 FOS.0.WT_JUN.4.T FOS.0.WT_JUN.2.TCC
#> 4 FOS.0.WT_JUN.58.A FOS.0.WT_JUN.20.ACC
#> 5 FOS.0.WT_JUN.88.A_JUN.89.G_JUN.90.G FOS.0.WT_JUN.30.AGG
#> 6 FOS.0.WT_JUN.89.G_JUN.90.G FOS.0.WT_JUN.30.GGG
#> mutantNameBaseHGVS mutantNameAA mutantNameAAHGVS
#> 1 FOS:c_JUN:c.37_39delinsCCC FOS.0.WT_JUN.13.P FOS:p_JUN:p.(Ala13Pro)
#> 2 FOS:c_JUN:c.37_39delinsCTC FOS.0.WT_JUN.13.L FOS:p_JUN:p.(Ala13Leu)
#> 3 FOS:c_JUN:c.4G>T FOS.0.WT_JUN.2.S FOS:p_JUN:p.(Ala2Ser)
#> 4 FOS:c_JUN:c.58T>A FOS.0.WT_JUN.20.T FOS:p_JUN:p.(Ser20Thr)
#> 5 FOS:c_JUN:c.88_90delinsAGG FOS.0.WT_JUN.30.R FOS:p_JUN:p.(Ala30Arg)
#> 6 FOS:c_JUN:c.89_90delinsGG FOS.0.WT_JUN.30.G FOS:p_JUN:p.(Ala30Gly)
#> mutationTypes
#> 1 nonsynonymous
#> 2 nonsynonymous
#> 3 nonsynonymous
#> 4 nonsynonymous
#> 5 nonsynonymous
#> 6 nonsynonymous
#> sequenceAA
#> 1 TDTLQAETDQLEDEKSALQTEIANLLKEKEKL_IARLEEKVKTLKPQNSELASTANMLREQVAQL
#> 2 TDTLQAETDQLEDEKSALQTEIANLLKEKEKL_IARLEEKVKTLKLQNSELASTANMLREQVAQL
#> 3 TDTLQAETDQLEDEKSALQTEIANLLKEKEKL_ISRLEEKVKTLKAQNSELASTANMLREQVAQL
#> 4 TDTLQAETDQLEDEKSALQTEIANLLKEKEKL_IARLEEKVKTLKAQNSELATTANMLREQVAQL
#> 5 TDTLQAETDQLEDEKSALQTEIANLLKEKEKL_IARLEEKVKTLKAQNSELASTANMLREQVRQL
#> 6 TDTLQAETDQLEDEKSALQTEIANLLKEKEKL_IARLEEKVKTLKAQNSELASTANMLREQVGQLFinally, the errorStatistics data.frame lists the number
of matching and mismatching bases in the constant sequences, stratified
by the phred quality score (from 0 to 99).
transInput$errorStatistics[rowSums(transInput$errorStatistics[, -1]) != 0, ]
#> PhredQuality nbrMatchForward nbrMismatchForward nbrMatchReverse
#> 15 14 160 11 204
#> 23 22 52 0 14
#> 28 27 302 4 474
#> 34 33 472 0 462
#> 38 37 4020 1 4405
#> nbrMismatchReverse
#> 15 17
#> 23 0
#> 28 3
#> 34 1
#> 38 0The errorStatistics output can be used to estimate the
sequencing error rate:
(propErrorsConstantF <- sum(transInput$errorStatistics$nbrMismatchForward) /
(nchar(transInput$parameters$constantForward) *
transInput$filterSummary$nbrRetained))
#> [1] 0.003185982
(propErrorsConstantR <- sum(transInput$errorStatistics$nbrMismatchReverse) /
(nchar(transInput$parameters$constantReverse) *
transInput$filterSummary$nbrRetained))
#> [1] 0.003763441Processing CIS data
Next, we illustrate the processing of the provided CIS experiment
example data. Recall that in this case, both the forward and reverse
variable sequence correspond to the same mutated FOS sequence,
and before matching to the wild type sequence, the two components need
to be merged to generate a single variable sequence (this is specified
via the mergeForwardReverse argument). As for the TRANS
data, we filter out reads with any adapter match, an average phred
quality below 20, any Ns in the UMI or variable sequence, more than one
mutated codon, or a mutated codon ending with A or T (represented by the
NNW value of the forbiddenMutatedCodonsForward
argument). Since the forward and reverse variable sequences are merged
into one variable sequence, only one wild type sequence is provided (the
wildTypeReverse argument will be ignored if specified).
cisInput <- digestFastqs(
fastqForward = file.path(datadir, "cisInput_1.fastq.gz"),
fastqReverse = file.path(datadir, "cisInput_2.fastq.gz"),
mergeForwardReverse = TRUE,
minOverlap = 0,
maxOverlap = 0,
maxFracMismatchOverlap = 1,
greedyOverlap = TRUE,
revComplForward = FALSE,
revComplReverse = TRUE,
adapterForward = "GGAAGAGCACACGTC",
adapterReverse = "GGAAGAGCGTCGTGT",
elementsForward = "SUCV",
elementsReverse = "SUCVS",
elementLengthsForward = c(1, 10, 18, 96),
elementLengthsReverse = c(1, 7, 17, 96, -1),
constantForward = "AACCGGAGGAGGGAGCTG",
constantReverse = "GAGTTCATCCTGGCAGC",
primerForward = "",
primerReverse = "",
avePhredMinForward = 20,
avePhredMinReverse = 20,
variableNMaxForward = 0,
variableNMaxReverse = 0,
umiNMax = 0,
wildTypeForward = c(FOS = "ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA"),
wildTypeReverse = "",
nbrMutatedCodonsMaxForward = 1,
nbrMutatedCodonsMaxReverse = 1,
forbiddenMutatedCodonsForward = "NNW",
forbiddenMutatedCodonsReverse = "NNW",
mutNameDelimiter = ".",
constantMaxDistForward = -1,
constantMaxDistReverse = -1,
verbose = TRUE
)
#> done enumerating forbidden codons (32)
#> done enumerating forbidden codons (32)
#> start reading sequences for file pair 1 of 1...
#> 1000 read pairs processed (16.7% retained)
#> done reading sequences
#> retained 67 unique features
cisInput$parameters
#> $fastqForward
#> [1] "/Users/runner/work/_temp/Library/mutscan/extdata/cisInput_1.fastq.gz"
#>
#> $fastqReverse
#> [1] "/Users/runner/work/_temp/Library/mutscan/extdata/cisInput_2.fastq.gz"
#>
#> $mergeForwardReverse
#> [1] TRUE
#>
#> $minOverlap
#> [1] 0
#>
#> $maxOverlap
#> [1] 0
#>
#> $minMergedLength
#> [1] 0
#>
#> $maxMergedLength
#> [1] 0
#>
#> $maxFracMismatchOverlap
#> [1] 1
#>
#> $greedyOverlap
#> [1] TRUE
#>
#> $revComplForward
#> [1] FALSE
#>
#> $revComplReverse
#> [1] TRUE
#>
#> $elementsForward
#> [1] "SUCV"
#>
#> $elementLengthsForward
#> [1] 1 10 18 96
#>
#> $elementsReverse
#> [1] "SUCVS"
#>
#> $elementLengthsReverse
#> [1] 1 7 17 96 -1
#>
#> $adapterForward
#> [1] "GGAAGAGCACACGTC"
#>
#> $adapterReverse
#> [1] "GGAAGAGCGTCGTGT"
#>
#> $primerForward
#> [1] ""
#>
#> $primerReverse
#> [1] ""
#>
#> $wildTypeForward
#> FOS
#> "ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA"
#>
#> $wildTypeReverse
#> r
#> ""
#>
#> $constantForward
#> [1] "AACCGGAGGAGGGAGCTG"
#>
#> $constantReverse
#> [1] "GAGTTCATCCTGGCAGC"
#>
#> $avePhredMinForward
#> [1] 20
#>
#> $avePhredMinReverse
#> [1] 20
#>
#> $variableNMaxForward
#> [1] 0
#>
#> $variableNMaxReverse
#> [1] 0
#>
#> $umiNMax
#> [1] 0
#>
#> $nbrMutatedCodonsMaxForward
#> [1] 1
#>
#> $nbrMutatedCodonsMaxReverse
#> [1] 1
#>
#> $nbrMutatedBasesMaxForward
#> [1] -1
#>
#> $nbrMutatedBasesMaxReverse
#> [1] -1
#>
#> $forbiddenMutatedCodonsForward
#> [1] "AAA" "AAT" "ACA" "ACT" "AGA" "AGT" "ATA" "ATT" "CAA" "CAT" "CCA" "CCT"
#> [13] "CGA" "CGT" "CTA" "CTT" "GAA" "GAT" "GCA" "GCT" "GGA" "GGT" "GTA" "GTT"
#> [25] "TAA" "TAT" "TCA" "TCT" "TGA" "TGT" "TTA" "TTT"
#>
#> $forbiddenMutatedCodonsReverse
#> [1] "AAA" "AAT" "ACA" "ACT" "AGA" "AGT" "ATA" "ATT" "CAA" "CAT" "CCA" "CCT"
#> [13] "CGA" "CGT" "CTA" "CTT" "GAA" "GAT" "GCA" "GCT" "GGA" "GGT" "GTA" "GTT"
#> [25] "TAA" "TAT" "TCA" "TCT" "TGA" "TGT" "TTA" "TTT"
#>
#> $useTreeWTmatch
#> [1] FALSE
#>
#> $collapseToWTForward
#> [1] FALSE
#>
#> $collapseToWTReverse
#> [1] FALSE
#>
#> $mutatedPhredMinForward
#> [1] 0
#>
#> $mutatedPhredMinReverse
#> [1] 0
#>
#> $mutNameDelimiter
#> [1] "."
#>
#> $constantMaxDistForward
#> [1] -1
#>
#> $constantMaxDistReverse
#> [1] -1
#>
#> $umiCollapseMaxDist
#> [1] 0
#>
#> $filteredReadsFastqForward
#> [1] ""
#>
#> $filteredReadsFastqReverse
#> [1] ""
#>
#> $maxNReads
#> [1] -1
#>
#> $nThreads
#> [1] 1
#>
#> $chunkSize
#> [1] 100000
#>
#> $maxReadLength
#> [1] 1024
#>
#> $processingInfo
#> [1] "Processed by mutscan v1.3.0 on 2026-04-28 18:01:11.165366"
cisInput$filterSummary
#> nbrTotal f1_nbrAdapter f2_nbrNoPrimer f3_nbrReadWrongLength
#> 1 1000 126 0 0
#> f4_nbrNoValidOverlap f5_nbrAvgVarQualTooLow f6_nbrTooManyNinVar
#> 1 0 0 44
#> f7_nbrTooManyNinUMI f8_nbrTooManyBestWTHits f9_nbrMutQualTooLow
#> 1 0 0 0
#> f10a_nbrTooManyMutCodons f10b_nbrTooManyMutBases f11_nbrForbiddenCodons
#> 1 581 0 82
#> f12_nbrTooManyMutConstant f13_nbrTooManyBestConstantHits nbrRetained
#> 1 0 0 167
cisInput$errorStatistics[rowSums(cisInput$errorStatistics[, -1]) != 0, ]
#> PhredQuality nbrMatchForward nbrMismatchForward nbrMatchReverse
#> 3 2 0 0 0
#> 15 14 51 1 41
#> 23 22 19 0 0
#> 28 27 79 1 56
#> 34 33 163 0 96
#> 38 37 2690 2 2620
#> nbrMismatchReverse
#> 3 4
#> 15 7
#> 23 0
#> 28 0
#> 34 1
#> 38 14The summaryTable now provides the number of reads and
unique UMIs observed for each variable sequence, and all values in the
mutNames column will start with FOS.
head(cisInput$summaryTable)
#> mutantName
#> 1 FOS.0.WT
#> 2 FOS.1.ACC
#> 3 FOS.1.ACG
#> 4 FOS.11.CTG
#> 5 FOS.13.GAC
#> 6 FOS.13.GAG
#> sequence
#> 1 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA
#> 2 ACCGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA
#> 3 ACGGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA
#> 4 ACTGATACACTCCAAGCGGAGACAGACCAACTGGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA
#> 5 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGACGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA
#> 6 ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGAGGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA
#> nbrReads maxNbrReads nbrUmis nbrMutBases nbrMutCodons nbrMutAAs varLengths
#> 1 77 77 77 0 0 0 96
#> 2 2 2 2 1 1 0 96
#> 3 1 1 1 1 1 0 96
#> 4 1 1 1 1 1 0 96
#> 5 1 1 1 1 1 0 96
#> 6 1 1 1 1 1 1 96
#> mutantNameBase mutantNameCodon mutantNameBaseHGVS mutantNameAA
#> 1 FOS.0.WT FOS.0.WT FOS:c FOS.0.WT
#> 2 FOS.3.C FOS.1.ACC FOS:c.3T>C FOS.0.WT
#> 3 FOS.3.G FOS.1.ACG FOS:c.3T>G FOS.0.WT
#> 4 FOS.33.G FOS.11.CTG FOS:c.33A>G FOS.0.WT
#> 5 FOS.39.C FOS.13.GAC FOS:c.39T>C FOS.0.WT
#> 6 FOS.39.G FOS.13.GAG FOS:c.39T>G FOS.13.E
#> mutantNameAAHGVS mutationTypes sequenceAA
#> 1 FOS:p TDTLQAETDQLEDEKSALQTEIANLLKEKEKL
#> 2 FOS:p silent TDTLQAETDQLEDEKSALQTEIANLLKEKEKL
#> 3 FOS:p silent TDTLQAETDQLEDEKSALQTEIANLLKEKEKL
#> 4 FOS:p silent TDTLQAETDQLEDEKSALQTEIANLLKEKEKL
#> 5 FOS:p silent TDTLQAETDQLEDEKSALQTEIANLLKEKEKL
#> 6 FOS:p.(Asp13Glu) nonsynonymous TDTLQAETDQLEEEKSALQTEIANLLKEKEKLProcessing TRANS data with primers
Finally, we illustrate the processing of the provided example data,
where the first read corresponds to one of 46 leucine zipper sequences,
and the second read is a mutated JUN sequence. We first need to
define the possible sequences for the forward read. If multiple wild
type sequences are provided like here, mutscan will match
each read against all of them and find the most similar one for each
read.
leu <- c(
ATF2 = "GATCCTGATGAAAAAAGGAGAAAGTTTTTAGAGCGAAATAGAGCAGCAGCTTCAAGATGCCGACAAAAAAGGAAAGTCTGGGTTCAGTCTTTAGAGAAGAAAGCTGAAGACTTGAGTTCATTAAATGGTCAGCTGCAGAGTGAAGTCACCCTGCTGAGAAATGAAGTGGCACAGCTGAAACAGCTTCTTCTGGCT",
ATF7 = "GATCCAGATGAGCGACGGCAGCGCTTTCTGGAGCGCAACCGGGCTGCAGCCTCCCGCTGCCGCCAAAAGCGAAAGCTGTGGGTGTCCTCCCTAGAGAAGAAGGCCGAAGAACTCACTTCTCAGAACATTCAGCTGAGTAATGAAGTCACATTACTACGCAATGAGGTGGCCCAGTTGAAACAGCTACTGTTAGCT",
CREB5 = "GATCCGGACGAGAGGCGGCGGAAATTTCTGGAACGGAACCGGGCAGCTGCCACCCGCTGCAGACAGAAGAGGAAGGTCTGGGTGATGTCATTGGAAAAGAAAGCAGAAGAACTCACCCAGACAAACATGCAGCTTCAGAATGAAGTGTCTATGTTGAAAAATGAGGTGGCCCAGCTGAAACAGTTGTTGTTAACA",
ATF3 = "GAAGAAGATGAAAGGAAAAAGAGGCGACGAGAAAGAAATAAGATTGCAGCTGCAAAGTGCCGAAACAAGAAGAAGGAGAAGACGGAGTGCCTGCAGAAAGAGTCGGAGAAGCTGGAAAGTGTGAATGCTGAACTGAAGGCTCAGATTGAGGAGCTCAAGAACGAGAAGCAGCATTTGATATACATGCTCAACCTT",
JDP2 = "GAGGAAGAGGAGCGAAGGAAAAGGCGCCGGGAGAAGAACAAAGTCGCAGCAGCCCGATGCCGGAACAAGAAGAAGGAGCGCACGGAGTTTCTGCAGCGGGAATCCGAGCGGCTGGAACTCATGAACGCAGAGCTGAAGACCCAGATTGAGGAGCTGAAGCAGGAGCGGCAGCAGCTCATCCTGATGCTGAACCGA",
ATF4 = "GAGAAACTGGATAAGAAGCTGAAAAAAATGGAGCAAAACAAGACAGCAGCCACTAGGTACCGCCAGAAGAAGAGGGCGGAGCAGGAGGCTCTTACTGGTGAGTGCAAAGAGCTGGAAAAGAAGAACGAGGCTCTAAAAGAGAGGGCGGATTCCCTGGCCAAGGAGATCCAGTACCTGAAAGATTTGATAGAAGAG",
ATF5 = "ACCCGAGGGGACCGCAAGCAAAAGAAGAGAGACCAGAACAAGTCGGCGGCTCTGAGGTACCGCCAGCGGAAGCGGGCAGAGGGTGAGGCCCTGGAGGGCGAGTGCCAGGGGCTGGAGGCACGGAATCGCGAGCTGAAGGAACGGGCAGAGTCCGTGGAGCGCGAGATCCAGTACGTCAAGGACCTGCTCATCGAG",
CREBZF = "AGTCCCCGGAAGGCGGCGGCGGCCGCTGCCCGCCTTAATCGACTGAAGAAGAAGGAGTACGTGATGGGGCTGGAGAGTCGAGTCCGGGGTCTGGCAGCCGAGAACCAGGAGCTGCGGGCCGAGAATCGGGAGCTGGGCAAACGCGTACAGGCACTGCAGGAGGAGAGTCGCTACCTACGGGCAGTCTTAGCCAAC",
BATF2 = "CCCAAGGAGCAACAAAGGCAGCTGAAGAAGCAGAAGAACCGGGCAGCCGCCCAGCGAAGCCGGCAGAAGCACACAGACAAGGCAGACGCCCTGCACCAGCAGCACGAGTCTCTGGAAAAAGACAACCTCGCCCTGCGGAAGGAGATCCAGTCCCTGCAGGCCGAGCTGGCGTGGTGGAGCCGGACCCTGCACGTG",
BATF3 = "GAGGATGATGACAGGAAGGTCCGAAGGAGAGAAAAAAACCGAGTTGCTGCTCAGAGAAGTCGGAAGAAGCAGACCCAGAAGGCTGACAAGCTCCATGAGGAATATGAGAGCCTGGAGCAAGAAAACACCATGCTGCGGAGAGAGATCGGGAAGCTGACAGAGGAGCTGAAGCACCTGACAGAGGCACTGAAGGAG",
CEBPE = "AAAGATAGCCTTGAGTACCGGCTGAGGCGGGAGCGCAACAACATCGCCGTGCGCAAGAGCCGAGACAAGGCCAAGAGGCGCATTCTGGAGACGCAGCAGAAGGTGCTGGAGTACATGGCAGAGAACGAGCGCCTCCGCAGCCGCGTGGAGCAGCTCACCCAGGAGCTAGACACCCTCCGCAACCTCTTCCGCCAG",
BACH1 = "CTGGATTGTATCCATGATATTCGAAGAAGAAGTAAAAACAGAATTGCTGCACAGCGCTGTCGCAAGAGAAAACTTGACTGTATACAGAATCTTGAATCAGAAATTGAGAAGCTGCAAAGTGAAAAGGAGAGCTTGTTGAAGGAAAGAGATCACATTTTGTCAACTCTGGGTGAGACAAAGCAGAACCTAACTGGA",
BACH2 = "TTAGAGTTTATTCATGATGTCCGACGGCGCAGCAAGAACCGCATCGCGGCCCAGCGCTGCCGCAAAAGGAAACTGGACTGTATTCAGAATTTAGAATGTGAAATCCGCAAATTGGTGTGTGAGAAAGAGAAACTGTTGTCAGAGAGGAATCAACTGAAAGCATGCATGGGGGAACTGTTGGACAACTTCTCCTGC",
NFE2L1 = "CTGAGCCTCATCCGAGACATCCGGCGCCGGGGCAAGAACAAGATGGCGGCGCAGAACTGCCGCAAGCGCAAGCTGGACACCATCCTGAATCTGGAGCGTGATGTGGAGGACCTGCAGCGTGACAAAGCCCGGCTGCTGCGGGAGAAAGTGGAGTTCCTGCGCTCCCTGCGACAGATGAAGCAGAAGGTCCAGAGC",
NFE2 = "CTAGCGCTAGTCCGGGACATCCGACGACGGGGCAAAAACAAGGTGGCAGCCCAGAACTGCCGCAAGAGGAAGCTGGAAACCATTGTGCAGCTGGAGCGGGAGCTGGAGCGGCTGACCAATGAACGGGAGCGGCTTCTCAGGGCCCGCGGGGAGGCAGACCGGACCCTGGAGGTCATGCGCCAACAGCTGACAGAG",
NFIL3 = "AAGAAAGATGCTATGTATTGGGAAAAAAGGCGGAAAAATAATGAAGCTGCCAAAAGATCTCGTGAGAAGCGTCGACTGAATGACCTGGTTTTAGAGAACAAACTAATTGCACTGGGAGAAGAAAACGCCACTTTAAAAGCTGAGCTGCTTTCACTAAAATTAAAGTTTGGTTTAATTAGCTCCACAGCATATGCT",
FOS = "GAAGAAGAAGAGAAAAGGAGAATCCGAAGGGAAAGGAATAAGATGGCTGCAGCCAAATGCCGCAACCGGAGGAGGGAGCTGACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTAGAGTTCATCCTGGCAGCT",
FOSB = "GAGGAAGAGGAGAAGCGAAGGGTGCGCCGGGAACGAAATAAACTAGCAGCAGCTAAATGCAGGAACCGGCGGAGGGAGCTGACCGACCGACTCCAGGCGGAGACAGATCAGTTGGAGGAAGAAAAAGCAGAGCTGGAGTCGGAGATCGCCGAGCTCCAAAAGGAGAAGGAACGTCTGGAGTTTGTGCTGGTGGCC",
FOSL1 = "GAGGAAGAGGAGCGCCGCCGAGTAAGGCGCGAGCGGAACAAGCTGGCTGCGGCCAAGTGCAGGAACCGGAGGAAGGAACTGACCGACTTCCTGCAGGCGGAGACTGACAAACTGGAAGATGAGAAATCTGGGCTGCAGCGAGAGATTGAGGAGCTGCAGAAGCAGAAGGAGCGCCTAGAGCTGGTGCTGGAAGCC",
FOSL2 = "GAAGAGGAGGAGAAGCGTCGCATCCGGCGGGAGAGGAACAAGCTGGCTGCAGCCAAGTGCCGGAACCGACGCCGGGAGCTGACAGAGAAGCTGCAGGCGGAGACAGAGGAGCTGGAGGAGGAGAAGTCAGGCCTGCAGAAGGAGATTGCTGAGCTGCAGAAGGAGAAGGAGAAGCTGGAGTTCATGTTGGTGGCT",
MAFB = "GTGATCCGCCTGAAGCAGAAGCGGCGGACCCTGAAGAACCGGGGCTACGCCCAGTCTTGCAGGTATAAACGCGTCCAGCAGAAGCACCACCTGGAGAATGAGAAGACGCAGCTCATTCAGCAGGTGGAGCAGCTTAAGCAGGAGGTGTCCCGGCTGGCCCGCGAGAGAGACGCCTACAAGGTCAAGTGCGAGAAA",
JUN = "CAGGAGCGGATCAAGGCGGAGAGGAAGCGCATGAGGAACCGCATCGCTGCCTCCAAGTGCCGAAAAAGGAAGCTGGAGAGAATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTTAAACAGAAAGTCATGAAC",
JUNB = "CAAGAGCGCATCAAAGTGGAGCGCAAGCGGCTGCGGAACCGGCTGGCGGCCACCAAGTGCCGGAAGCGGAAGCTGGAGCGCATCGCGCGCCTGGAGGACAAGGTGAAGACGCTCAAGGCCGAGAACGCGGGGCTGTCGAGTACCGCCGGCCTCCTCCGGGAGCAGGTGGCCCAGCTCAAACAGAAGGTCATGACC",
JUND = "CAGGAGCGCATCAAGGCGGAGCGCAAGCGGCTGCGCAACCGCATCGCCGCCTCCAAGTGCCGCAAGCGCAAGCTGGAGCGCATCTCGCGCCTGGAAGAGAAAGTGAAGACCCTCAAGAGTCAGAACACGGAGCTGGCGTCCACGGCGAGCCTGCTGCGCGAGCAGGTGGCGCAGCTCAAGCAGAAAGTCCTCAGC",
CREB3 = "GAACAAATTCTGAAACGTGTGCGGAGGAAGATTCGAAATAAAAGATCTGCTCAAGAGAGCCGCAGGAAAAAGAAGGTGTATGTTGGGGGTTTAGAGAGCAGGGTCTTGAAATACACAGCCCAGAATATGGAGCTTCAGAACAAAGTACAGCTTCTGGAGGAACAGAATTTGTCCCTTCTAGATCAACTGAGGAAA",
HLF = "CTGAAGGATGACAAGTACTGGGCAAGGCGCAGAAAGAACAACATGGCAGCCAAGCGCTCCCGCGACGCCCGGAGGCTGAAAGAGAACCAGATCGCCATCCGGGCCTCGTTCCTGGAGAAGGAGAACTCGGCCCTCCGCCAGGAGGTGGCTGACTTGAGGAAGGAGCTGGGCAAATGCAAGAACATACTTGCCAAG",
MAFG = "ATCGTCCAGCTGAAGCAGCGCCGGCGCACGCTCAAGAACCGCGGCTACGCTGCCAGCTGCCGCGTGAAGCGGGTGACGCAGAAGGAGGAGCTGGAGAAGCAGAAGGCGGAGCTGCAGCAGGAGGTGGAGAAGCTGGCCTCAGAGAACGCCAGCATGAAGCTGGAGCTCGACGCGCTGCGCTCCAAGTACGAGGCG",
MAFK = "GTGACCCGCCTGAAGCAGCGTCGGCGCACACTCAAGAACCGCGGCTACGCGGCCAGCTGCCGCATCAAGCGGGTGACGCAGAAGGAGGAGCTGGAGCGGCAGCGCGTGGAGCTGCAGCAGGAGGTGGAGAAGCTGGCGCGTGAGAACAGCAGCATGCGGCTGGAGCTGGACGCCCTGCGCTCCAAGTACGAGGCG",
XBP1 = "AGCCCCGAGGAGAAGGCGCTGAGGAGGAAACTGAAAAACAGAGTAGCAGCTCAGACTGCCAGAGATCGAAAGAAGGCTCGAATGAGTGAGCTGGAACAGCAAGTGGTAGATTTAGAAGAAGAGAACCAAAAACTTTTGCTAGAAAATCAGCTTTTACGAGAGAAAACTCATGGCCTTGTAGTTGAGAACCAGGAG",
ATF6 = "ATTGCTGTGCTAAGGAGACAGCAACGTATGATAAAAAATCGAGAATCCGCTTGTCAGTCTCGCAAGAAGAAGAAAGAATATATGCTAGGGTTAGAGGCGAGATTAAAGGCTGCCCTCTCAGAAAACGAGCAACTGAAGAAAGAAAATGGAACACTGAAGCGGCAGCTGGATGAAGTTGTGTCAGAGAACCAGAGG",
ATF6B = "GCAAAGCTGCTGAAGCGGCAGCAGCGAATGATCAAGAACCGGGAGTCAGCCTGCCAGTCCCGGAGAAAGAAGAAAGAGTATCTGCAGGGACTGGAGGCTCGGCTGCAAGCAGTACTGGCTGACAACCAGCAGCTCCGCCGAGAGAATGCTGCCCTCCGGCGGCGGCTGGAGGCCCTGCTGGCTGAAAACAGCGAG",
CEBPA = "AAGAACAGCAACGAGTACCGGGTGCGGCGCGAGCGCAACAACATCGCGGTGCGCAAGAGCCGCGACAAGGCCAAGCAGCGCAACGTGGAGACGCAGCAGAAGGTGCTGGAGCTGACCAGTGACAATGACCGCCTGCGCAAGCGGGTGGAACAGCTGAGCCGCGAACTGGACACGCTGCGGGGCATCTTCCGCCAG",
CEBPB = "AAGCACAGCGACGAGTACAAGATCCGGCGCGAGCGCAACAACATCGCCGTGCGCAAGAGCCGCGACAAGGCCAAGATGCGCAACCTGGAGACGCAGCACAAGGTCCTGGAGCTCACGGCCGAGAACGAGCGGCTGCAGAAGAAGGTGGAGCAGCTGTCGCGCGAGCTCAGCACCCTGCGGAACTTGTTCAAGCAG",
CEBPD = "CGCGGCAGCCCCGAGTACCGGCAGCGGCGCGAGCGCAACAACATCGCCGTGCGCAAGAGCCGCGACAAGGCCAAGCGGCGCAACCAGGAGATGCAGCAGAAGTTGGTGGAGCTGTCGGCTGAGAACGAGAAGCTGCACCAGCGCGTGGAGCAGCTCACGCGGGACCTGGCCGGCCTCCGGCAGTTCTTCAAGCAG",
CEBPG = "CGAAACAGTGACGAGTATCGGCAACGCCGAGAGAGGAACAACATGGCTGTGAAAAAGAGCCGGTTGAAAAGCAAGCAGAAAGCACAAGACACACTGCAGAGAGTCAATCAGCTCAAAGAAGAGAATGAACGGTTGGAAGCAAAAATCAAATTGCTGACCAAGGAATTAAGTGTACTCAAAGATTTGTTTCTTGAG",
CREB1 = "GAAGCAGCACGAAAGAGAGAGGTCCGTCTAATGAAGAACAGGGAAGCAGCTCGAGAGTGTCGTAGAAAGAAGAAAGAATATGTGAAATGTTTAGAAAACAGAGTGGCAGTGCTTGAAAATCAAAACAAGACATTGATTGAGGAGCTAAAAGCACTTAAGGACCTTTACTGCCACAAATCAGAT",
CREB3L1 = "GAGAAGGCCTTGAAGAGAGTCCGGAGGAAAATCAAGAACAAGATCTCAGCCCAGGAGAGCCGTCGTAAGAAGAAGGAGTATGTGGAGTGTCTAGAAAAGAAGGTGGAGACATTTACATCTGAGAACAATGAACTGTGGAAGAAGGTGGAGACCCTGGAGAATGCCAACAGGACCCTGCTCCAGCAGCTGCAGAAA",
CREB3L2 = "GAGAAGGCCCTGAAGAAAATTCGGAGGAAGATCAAGAATAAGATTTCTGCTCAGGAAAGTAGGAGAAAGAAGAAAGAATACATGGACAGCCTGGAGAAAAAAGTGGAGTCTTGTTCAACTGAGAACTTGGAGCTTCGGAAGAAGGTAGAGGTTCTAGAGAACACTAATAGGACTCTCCTTCAGCAACTCCAGAAG",
CREB3L3 = "GAGCGAGTGCTGAAAAAAATCCGCCGGAAAATCCGGAACAAGCAGTCGGCGCAAGAAAGCAGGAAGAAGAAGAAGGAATATATCGATGGCCTGGAGACTCGGATGTCAGCTTGCACTGCTCAGAATCAGGAGTTACAGAGGAAAGTCTTGCATCTCGAGAAGCAAAACCTGTCCCTCTTGGAGCAACTGAAGAAA",
CREB3L4 = "GAGAGGGTCCTCAAGAAGGTCAGGAGGAAAATCCGTAACAAGCAGTCAGCTCAGGACAGTCGGCGGCGGAAGAAGGAGTACATTGATGGGCTGGAGAGCAGGGTGGCAGCCTGTTCTGCACAGAACCAAGAATTACAGAAAAAAGTCCAGGAGCTGGAGAGGCACAACATCTCCTTGGTAGCTCAGCTCCGCCAG",
CREBL2 = "CCAGCCAAAATTGACTTGAAAGCAAAACTTGAGAGGAGCCGGCAGAGTGCAAGAGAATGCCGAGCCCGAAAAAAGCTGAGATATCAGTATTTGGAAGAGTTGGTATCCAGTCGAGAAAGAGCTATATGTGCCCTCAGAGAGGAACTGGAAATGTACAAGCAGTGGTGCATGGCAATGGACCAAGGAAAAATCCCT",
CREBRF = "CCCTTAACAGCCCGACCAAGGTCAAGGAAGGAAAAAAATAAGCTGGCTTCCAGAGCTTGTCGGTTAAAGAAGAAAGCCCAGTATGAAGCTAATAAAGTGAAATTATGGGGCCTCAACACAGAATATGATAATTTATTGTTTGTAATCAACTCCATCAAGCAAGAGATTGTAAACCGGGTACAGAATCCAAGAGAT",
DBP = "CAGAAGGATGAGAAATACTGGAGCCGGCGGTACAAGAACAACGAGGCAGCCAAGCGGTCCCGTGACGCCCGGCGGCTCAAGGAGAACCAGATATCGGTGCGGGCGGCCTTCCTGGAGAAGGAGAACGCCCTGCTGCGGCAGGAAGTTGTGGCCGTGCGCCAGGAGCTGTCCCACTACCGCGCCGTGCTGTCCCGA",
NFE2L2 = "CTTGCATTAATTCGGGATATACGTAGGAGGGGTAAGAATAAAGTGGCTGCTCAGAATTGCAGAAAAAGAAAACTGGAAAATATAGTAGAACTAGAGCAAGATTTAGATCATTTGAAAGATGAAAAAGAAAAATTGCTCAAAGAAAAAGGAGAAAATGACAAAAGCCTTCACCTACTGAAAAAACAACTCAGCACC",
NFE2L3 = "GTCTCACTTATCCGTGACATCAGACGAAGAGGGAAAAATAAAGTTGCTGCGCAGAACTGTCGTAAACGCAAATTGGACATAATTTTGAATTTAGAAGATGATGTATGTAACTTGCAAGCAAAGAAGGAAACTCTTAAGAGAGAGCAAGCACAATGTAACAAAGCTATTAACATAATGAAACAGAAACTGCATGAC",
TEF = "CAGAAGGATGAAAAGTACTGGACAAGACGCAAGAAGAACAACGTGGCAGCTAAACGGTCACGGGATGCCCGGCGCCTGAAAGAGAATCAGATCACCATCCGGGCAGCCTTCCTGGAGAAGGAGAACACAGCCCTGCGGACGGAGGTGGCCGAGCTACGCAAGGAGGTGGGCAAGTGCAAGACCATCGTGTCCAAG"
)Next we process the data, not allowing any mismatches in the forward read, but 1 mismatching codon in the reverse read. Now, we assume that the variable sequence starts immediately after the provided primers, and hence we don’t specify any UMI/constant sequence lengths. For the forward read, the variable region is taken to be the remainder of the read (after the primer), whereas for the reverse read, we specify the variable sequence length to 96.
leujunt0 <- digestFastqs(
fastqForward = file.path(datadir, "leujunt0_1.fastq.gz"),
fastqReverse = file.path(datadir, "leujunt0_2.fastq.gz"),
mergeForwardReverse = FALSE,
revComplForward = FALSE,
revComplReverse = FALSE,
elementsForward = "SPV",
elementsReverse = "SPVS",
elementLengthsForward = c(-1, 19, -1),
elementLengthsReverse = c(-1, 20, 96, -1),
constantForward = "",
constantReverse = "",
adapterForward = "",
adapterReverse = "",
primerForward = "GTCAGGTGGAGGCGGATCC",
primerReverse = "GAAAAAGGAAGCTGGAGAGA",
avePhredMinForward = 20,
avePhredMinReverse = 20,
variableNMaxForward = 0,
variableNMaxReverse = 0,
umiNMax = 0,
wildTypeForward = leu,
wildTypeReverse = c(JUN = "ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT"),
nbrMutatedCodonsMaxForward = 0,
nbrMutatedCodonsMaxReverse = 1,
forbiddenMutatedCodonsForward = "",
forbiddenMutatedCodonsReverse = "NNW",
mutatedPhredMinForward = 0.0,
mutatedPhredMinReverse = 0.0,
mutNameDelimiter = ".",
verbose = TRUE
)
#> done enumerating forbidden codons (0)
#> done enumerating forbidden codons (32)
#> start reading sequences for file pair 1 of 1...
#> 1000 read pairs processed (60% retained)
#> done reading sequences
#> retained 587 unique features
leujunt0$parameters
#> $fastqForward
#> [1] "/Users/runner/work/_temp/Library/mutscan/extdata/leujunt0_1.fastq.gz"
#>
#> $fastqReverse
#> [1] "/Users/runner/work/_temp/Library/mutscan/extdata/leujunt0_2.fastq.gz"
#>
#> $mergeForwardReverse
#> [1] FALSE
#>
#> $minOverlap
#> [1] 0
#>
#> $maxOverlap
#> [1] 0
#>
#> $minMergedLength
#> [1] 0
#>
#> $maxMergedLength
#> [1] 0
#>
#> $maxFracMismatchOverlap
#> [1] 1
#>
#> $greedyOverlap
#> [1] TRUE
#>
#> $revComplForward
#> [1] FALSE
#>
#> $revComplReverse
#> [1] FALSE
#>
#> $elementsForward
#> [1] "SPV"
#>
#> $elementLengthsForward
#> [1] -1 19 -1
#>
#> $elementsReverse
#> [1] "SPVS"
#>
#> $elementLengthsReverse
#> [1] -1 20 96 -1
#>
#> $adapterForward
#> [1] ""
#>
#> $adapterReverse
#> [1] ""
#>
#> $primerForward
#> [1] "GTCAGGTGGAGGCGGATCC"
#>
#> $primerReverse
#> [1] "GAAAAAGGAAGCTGGAGAGA"
#>
#> $wildTypeForward
#> ATF2
#> "GATCCTGATGAAAAAAGGAGAAAGTTTTTAGAGCGAAATAGAGCAGCAGCTTCAAGATGCCGACAAAAAAGGAAAGTCTGGGTTCAGTCTTTAGAGAAGAAAGCTGAAGACTTGAGTTCATTAAATGGTCAGCTGCAGAGTGAAGTCACCCTGCTGAGAAATGAAGTGGCACAGCTGAAACAGCTTCTTCTGGCT"
#> ATF7
#> "GATCCAGATGAGCGACGGCAGCGCTTTCTGGAGCGCAACCGGGCTGCAGCCTCCCGCTGCCGCCAAAAGCGAAAGCTGTGGGTGTCCTCCCTAGAGAAGAAGGCCGAAGAACTCACTTCTCAGAACATTCAGCTGAGTAATGAAGTCACATTACTACGCAATGAGGTGGCCCAGTTGAAACAGCTACTGTTAGCT"
#> CREB5
#> "GATCCGGACGAGAGGCGGCGGAAATTTCTGGAACGGAACCGGGCAGCTGCCACCCGCTGCAGACAGAAGAGGAAGGTCTGGGTGATGTCATTGGAAAAGAAAGCAGAAGAACTCACCCAGACAAACATGCAGCTTCAGAATGAAGTGTCTATGTTGAAAAATGAGGTGGCCCAGCTGAAACAGTTGTTGTTAACA"
#> ATF3
#> "GAAGAAGATGAAAGGAAAAAGAGGCGACGAGAAAGAAATAAGATTGCAGCTGCAAAGTGCCGAAACAAGAAGAAGGAGAAGACGGAGTGCCTGCAGAAAGAGTCGGAGAAGCTGGAAAGTGTGAATGCTGAACTGAAGGCTCAGATTGAGGAGCTCAAGAACGAGAAGCAGCATTTGATATACATGCTCAACCTT"
#> JDP2
#> "GAGGAAGAGGAGCGAAGGAAAAGGCGCCGGGAGAAGAACAAAGTCGCAGCAGCCCGATGCCGGAACAAGAAGAAGGAGCGCACGGAGTTTCTGCAGCGGGAATCCGAGCGGCTGGAACTCATGAACGCAGAGCTGAAGACCCAGATTGAGGAGCTGAAGCAGGAGCGGCAGCAGCTCATCCTGATGCTGAACCGA"
#> ATF4
#> "GAGAAACTGGATAAGAAGCTGAAAAAAATGGAGCAAAACAAGACAGCAGCCACTAGGTACCGCCAGAAGAAGAGGGCGGAGCAGGAGGCTCTTACTGGTGAGTGCAAAGAGCTGGAAAAGAAGAACGAGGCTCTAAAAGAGAGGGCGGATTCCCTGGCCAAGGAGATCCAGTACCTGAAAGATTTGATAGAAGAG"
#> ATF5
#> "ACCCGAGGGGACCGCAAGCAAAAGAAGAGAGACCAGAACAAGTCGGCGGCTCTGAGGTACCGCCAGCGGAAGCGGGCAGAGGGTGAGGCCCTGGAGGGCGAGTGCCAGGGGCTGGAGGCACGGAATCGCGAGCTGAAGGAACGGGCAGAGTCCGTGGAGCGCGAGATCCAGTACGTCAAGGACCTGCTCATCGAG"
#> CREBZF
#> "AGTCCCCGGAAGGCGGCGGCGGCCGCTGCCCGCCTTAATCGACTGAAGAAGAAGGAGTACGTGATGGGGCTGGAGAGTCGAGTCCGGGGTCTGGCAGCCGAGAACCAGGAGCTGCGGGCCGAGAATCGGGAGCTGGGCAAACGCGTACAGGCACTGCAGGAGGAGAGTCGCTACCTACGGGCAGTCTTAGCCAAC"
#> BATF2
#> "CCCAAGGAGCAACAAAGGCAGCTGAAGAAGCAGAAGAACCGGGCAGCCGCCCAGCGAAGCCGGCAGAAGCACACAGACAAGGCAGACGCCCTGCACCAGCAGCACGAGTCTCTGGAAAAAGACAACCTCGCCCTGCGGAAGGAGATCCAGTCCCTGCAGGCCGAGCTGGCGTGGTGGAGCCGGACCCTGCACGTG"
#> BATF3
#> "GAGGATGATGACAGGAAGGTCCGAAGGAGAGAAAAAAACCGAGTTGCTGCTCAGAGAAGTCGGAAGAAGCAGACCCAGAAGGCTGACAAGCTCCATGAGGAATATGAGAGCCTGGAGCAAGAAAACACCATGCTGCGGAGAGAGATCGGGAAGCTGACAGAGGAGCTGAAGCACCTGACAGAGGCACTGAAGGAG"
#> CEBPE
#> "AAAGATAGCCTTGAGTACCGGCTGAGGCGGGAGCGCAACAACATCGCCGTGCGCAAGAGCCGAGACAAGGCCAAGAGGCGCATTCTGGAGACGCAGCAGAAGGTGCTGGAGTACATGGCAGAGAACGAGCGCCTCCGCAGCCGCGTGGAGCAGCTCACCCAGGAGCTAGACACCCTCCGCAACCTCTTCCGCCAG"
#> BACH1
#> "CTGGATTGTATCCATGATATTCGAAGAAGAAGTAAAAACAGAATTGCTGCACAGCGCTGTCGCAAGAGAAAACTTGACTGTATACAGAATCTTGAATCAGAAATTGAGAAGCTGCAAAGTGAAAAGGAGAGCTTGTTGAAGGAAAGAGATCACATTTTGTCAACTCTGGGTGAGACAAAGCAGAACCTAACTGGA"
#> BACH2
#> "TTAGAGTTTATTCATGATGTCCGACGGCGCAGCAAGAACCGCATCGCGGCCCAGCGCTGCCGCAAAAGGAAACTGGACTGTATTCAGAATTTAGAATGTGAAATCCGCAAATTGGTGTGTGAGAAAGAGAAACTGTTGTCAGAGAGGAATCAACTGAAAGCATGCATGGGGGAACTGTTGGACAACTTCTCCTGC"
#> NFE2L1
#> "CTGAGCCTCATCCGAGACATCCGGCGCCGGGGCAAGAACAAGATGGCGGCGCAGAACTGCCGCAAGCGCAAGCTGGACACCATCCTGAATCTGGAGCGTGATGTGGAGGACCTGCAGCGTGACAAAGCCCGGCTGCTGCGGGAGAAAGTGGAGTTCCTGCGCTCCCTGCGACAGATGAAGCAGAAGGTCCAGAGC"
#> NFE2
#> "CTAGCGCTAGTCCGGGACATCCGACGACGGGGCAAAAACAAGGTGGCAGCCCAGAACTGCCGCAAGAGGAAGCTGGAAACCATTGTGCAGCTGGAGCGGGAGCTGGAGCGGCTGACCAATGAACGGGAGCGGCTTCTCAGGGCCCGCGGGGAGGCAGACCGGACCCTGGAGGTCATGCGCCAACAGCTGACAGAG"
#> NFIL3
#> "AAGAAAGATGCTATGTATTGGGAAAAAAGGCGGAAAAATAATGAAGCTGCCAAAAGATCTCGTGAGAAGCGTCGACTGAATGACCTGGTTTTAGAGAACAAACTAATTGCACTGGGAGAAGAAAACGCCACTTTAAAAGCTGAGCTGCTTTCACTAAAATTAAAGTTTGGTTTAATTAGCTCCACAGCATATGCT"
#> FOS
#> "GAAGAAGAAGAGAAAAGGAGAATCCGAAGGGAAAGGAATAAGATGGCTGCAGCCAAATGCCGCAACCGGAGGAGGGAGCTGACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTAGAGTTCATCCTGGCAGCT"
#> FOSB
#> "GAGGAAGAGGAGAAGCGAAGGGTGCGCCGGGAACGAAATAAACTAGCAGCAGCTAAATGCAGGAACCGGCGGAGGGAGCTGACCGACCGACTCCAGGCGGAGACAGATCAGTTGGAGGAAGAAAAAGCAGAGCTGGAGTCGGAGATCGCCGAGCTCCAAAAGGAGAAGGAACGTCTGGAGTTTGTGCTGGTGGCC"
#> FOSL1
#> "GAGGAAGAGGAGCGCCGCCGAGTAAGGCGCGAGCGGAACAAGCTGGCTGCGGCCAAGTGCAGGAACCGGAGGAAGGAACTGACCGACTTCCTGCAGGCGGAGACTGACAAACTGGAAGATGAGAAATCTGGGCTGCAGCGAGAGATTGAGGAGCTGCAGAAGCAGAAGGAGCGCCTAGAGCTGGTGCTGGAAGCC"
#> FOSL2
#> "GAAGAGGAGGAGAAGCGTCGCATCCGGCGGGAGAGGAACAAGCTGGCTGCAGCCAAGTGCCGGAACCGACGCCGGGAGCTGACAGAGAAGCTGCAGGCGGAGACAGAGGAGCTGGAGGAGGAGAAGTCAGGCCTGCAGAAGGAGATTGCTGAGCTGCAGAAGGAGAAGGAGAAGCTGGAGTTCATGTTGGTGGCT"
#> MAFB
#> "GTGATCCGCCTGAAGCAGAAGCGGCGGACCCTGAAGAACCGGGGCTACGCCCAGTCTTGCAGGTATAAACGCGTCCAGCAGAAGCACCACCTGGAGAATGAGAAGACGCAGCTCATTCAGCAGGTGGAGCAGCTTAAGCAGGAGGTGTCCCGGCTGGCCCGCGAGAGAGACGCCTACAAGGTCAAGTGCGAGAAA"
#> JUN
#> "CAGGAGCGGATCAAGGCGGAGAGGAAGCGCATGAGGAACCGCATCGCTGCCTCCAAGTGCCGAAAAAGGAAGCTGGAGAGAATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTTAAACAGAAAGTCATGAAC"
#> JUNB
#> "CAAGAGCGCATCAAAGTGGAGCGCAAGCGGCTGCGGAACCGGCTGGCGGCCACCAAGTGCCGGAAGCGGAAGCTGGAGCGCATCGCGCGCCTGGAGGACAAGGTGAAGACGCTCAAGGCCGAGAACGCGGGGCTGTCGAGTACCGCCGGCCTCCTCCGGGAGCAGGTGGCCCAGCTCAAACAGAAGGTCATGACC"
#> JUND
#> "CAGGAGCGCATCAAGGCGGAGCGCAAGCGGCTGCGCAACCGCATCGCCGCCTCCAAGTGCCGCAAGCGCAAGCTGGAGCGCATCTCGCGCCTGGAAGAGAAAGTGAAGACCCTCAAGAGTCAGAACACGGAGCTGGCGTCCACGGCGAGCCTGCTGCGCGAGCAGGTGGCGCAGCTCAAGCAGAAAGTCCTCAGC"
#> CREB3
#> "GAACAAATTCTGAAACGTGTGCGGAGGAAGATTCGAAATAAAAGATCTGCTCAAGAGAGCCGCAGGAAAAAGAAGGTGTATGTTGGGGGTTTAGAGAGCAGGGTCTTGAAATACACAGCCCAGAATATGGAGCTTCAGAACAAAGTACAGCTTCTGGAGGAACAGAATTTGTCCCTTCTAGATCAACTGAGGAAA"
#> HLF
#> "CTGAAGGATGACAAGTACTGGGCAAGGCGCAGAAAGAACAACATGGCAGCCAAGCGCTCCCGCGACGCCCGGAGGCTGAAAGAGAACCAGATCGCCATCCGGGCCTCGTTCCTGGAGAAGGAGAACTCGGCCCTCCGCCAGGAGGTGGCTGACTTGAGGAAGGAGCTGGGCAAATGCAAGAACATACTTGCCAAG"
#> MAFG
#> "ATCGTCCAGCTGAAGCAGCGCCGGCGCACGCTCAAGAACCGCGGCTACGCTGCCAGCTGCCGCGTGAAGCGGGTGACGCAGAAGGAGGAGCTGGAGAAGCAGAAGGCGGAGCTGCAGCAGGAGGTGGAGAAGCTGGCCTCAGAGAACGCCAGCATGAAGCTGGAGCTCGACGCGCTGCGCTCCAAGTACGAGGCG"
#> MAFK
#> "GTGACCCGCCTGAAGCAGCGTCGGCGCACACTCAAGAACCGCGGCTACGCGGCCAGCTGCCGCATCAAGCGGGTGACGCAGAAGGAGGAGCTGGAGCGGCAGCGCGTGGAGCTGCAGCAGGAGGTGGAGAAGCTGGCGCGTGAGAACAGCAGCATGCGGCTGGAGCTGGACGCCCTGCGCTCCAAGTACGAGGCG"
#> XBP1
#> "AGCCCCGAGGAGAAGGCGCTGAGGAGGAAACTGAAAAACAGAGTAGCAGCTCAGACTGCCAGAGATCGAAAGAAGGCTCGAATGAGTGAGCTGGAACAGCAAGTGGTAGATTTAGAAGAAGAGAACCAAAAACTTTTGCTAGAAAATCAGCTTTTACGAGAGAAAACTCATGGCCTTGTAGTTGAGAACCAGGAG"
#> ATF6
#> "ATTGCTGTGCTAAGGAGACAGCAACGTATGATAAAAAATCGAGAATCCGCTTGTCAGTCTCGCAAGAAGAAGAAAGAATATATGCTAGGGTTAGAGGCGAGATTAAAGGCTGCCCTCTCAGAAAACGAGCAACTGAAGAAAGAAAATGGAACACTGAAGCGGCAGCTGGATGAAGTTGTGTCAGAGAACCAGAGG"
#> ATF6B
#> "GCAAAGCTGCTGAAGCGGCAGCAGCGAATGATCAAGAACCGGGAGTCAGCCTGCCAGTCCCGGAGAAAGAAGAAAGAGTATCTGCAGGGACTGGAGGCTCGGCTGCAAGCAGTACTGGCTGACAACCAGCAGCTCCGCCGAGAGAATGCTGCCCTCCGGCGGCGGCTGGAGGCCCTGCTGGCTGAAAACAGCGAG"
#> CEBPA
#> "AAGAACAGCAACGAGTACCGGGTGCGGCGCGAGCGCAACAACATCGCGGTGCGCAAGAGCCGCGACAAGGCCAAGCAGCGCAACGTGGAGACGCAGCAGAAGGTGCTGGAGCTGACCAGTGACAATGACCGCCTGCGCAAGCGGGTGGAACAGCTGAGCCGCGAACTGGACACGCTGCGGGGCATCTTCCGCCAG"
#> CEBPB
#> "AAGCACAGCGACGAGTACAAGATCCGGCGCGAGCGCAACAACATCGCCGTGCGCAAGAGCCGCGACAAGGCCAAGATGCGCAACCTGGAGACGCAGCACAAGGTCCTGGAGCTCACGGCCGAGAACGAGCGGCTGCAGAAGAAGGTGGAGCAGCTGTCGCGCGAGCTCAGCACCCTGCGGAACTTGTTCAAGCAG"
#> CEBPD
#> "CGCGGCAGCCCCGAGTACCGGCAGCGGCGCGAGCGCAACAACATCGCCGTGCGCAAGAGCCGCGACAAGGCCAAGCGGCGCAACCAGGAGATGCAGCAGAAGTTGGTGGAGCTGTCGGCTGAGAACGAGAAGCTGCACCAGCGCGTGGAGCAGCTCACGCGGGACCTGGCCGGCCTCCGGCAGTTCTTCAAGCAG"
#> CEBPG
#> "CGAAACAGTGACGAGTATCGGCAACGCCGAGAGAGGAACAACATGGCTGTGAAAAAGAGCCGGTTGAAAAGCAAGCAGAAAGCACAAGACACACTGCAGAGAGTCAATCAGCTCAAAGAAGAGAATGAACGGTTGGAAGCAAAAATCAAATTGCTGACCAAGGAATTAAGTGTACTCAAAGATTTGTTTCTTGAG"
#> CREB1
#> "GAAGCAGCACGAAAGAGAGAGGTCCGTCTAATGAAGAACAGGGAAGCAGCTCGAGAGTGTCGTAGAAAGAAGAAAGAATATGTGAAATGTTTAGAAAACAGAGTGGCAGTGCTTGAAAATCAAAACAAGACATTGATTGAGGAGCTAAAAGCACTTAAGGACCTTTACTGCCACAAATCAGAT"
#> CREB3L1
#> "GAGAAGGCCTTGAAGAGAGTCCGGAGGAAAATCAAGAACAAGATCTCAGCCCAGGAGAGCCGTCGTAAGAAGAAGGAGTATGTGGAGTGTCTAGAAAAGAAGGTGGAGACATTTACATCTGAGAACAATGAACTGTGGAAGAAGGTGGAGACCCTGGAGAATGCCAACAGGACCCTGCTCCAGCAGCTGCAGAAA"
#> CREB3L2
#> "GAGAAGGCCCTGAAGAAAATTCGGAGGAAGATCAAGAATAAGATTTCTGCTCAGGAAAGTAGGAGAAAGAAGAAAGAATACATGGACAGCCTGGAGAAAAAAGTGGAGTCTTGTTCAACTGAGAACTTGGAGCTTCGGAAGAAGGTAGAGGTTCTAGAGAACACTAATAGGACTCTCCTTCAGCAACTCCAGAAG"
#> CREB3L3
#> "GAGCGAGTGCTGAAAAAAATCCGCCGGAAAATCCGGAACAAGCAGTCGGCGCAAGAAAGCAGGAAGAAGAAGAAGGAATATATCGATGGCCTGGAGACTCGGATGTCAGCTTGCACTGCTCAGAATCAGGAGTTACAGAGGAAAGTCTTGCATCTCGAGAAGCAAAACCTGTCCCTCTTGGAGCAACTGAAGAAA"
#> CREB3L4
#> "GAGAGGGTCCTCAAGAAGGTCAGGAGGAAAATCCGTAACAAGCAGTCAGCTCAGGACAGTCGGCGGCGGAAGAAGGAGTACATTGATGGGCTGGAGAGCAGGGTGGCAGCCTGTTCTGCACAGAACCAAGAATTACAGAAAAAAGTCCAGGAGCTGGAGAGGCACAACATCTCCTTGGTAGCTCAGCTCCGCCAG"
#> CREBL2
#> "CCAGCCAAAATTGACTTGAAAGCAAAACTTGAGAGGAGCCGGCAGAGTGCAAGAGAATGCCGAGCCCGAAAAAAGCTGAGATATCAGTATTTGGAAGAGTTGGTATCCAGTCGAGAAAGAGCTATATGTGCCCTCAGAGAGGAACTGGAAATGTACAAGCAGTGGTGCATGGCAATGGACCAAGGAAAAATCCCT"
#> CREBRF
#> "CCCTTAACAGCCCGACCAAGGTCAAGGAAGGAAAAAAATAAGCTGGCTTCCAGAGCTTGTCGGTTAAAGAAGAAAGCCCAGTATGAAGCTAATAAAGTGAAATTATGGGGCCTCAACACAGAATATGATAATTTATTGTTTGTAATCAACTCCATCAAGCAAGAGATTGTAAACCGGGTACAGAATCCAAGAGAT"
#> DBP
#> "CAGAAGGATGAGAAATACTGGAGCCGGCGGTACAAGAACAACGAGGCAGCCAAGCGGTCCCGTGACGCCCGGCGGCTCAAGGAGAACCAGATATCGGTGCGGGCGGCCTTCCTGGAGAAGGAGAACGCCCTGCTGCGGCAGGAAGTTGTGGCCGTGCGCCAGGAGCTGTCCCACTACCGCGCCGTGCTGTCCCGA"
#> NFE2L2
#> "CTTGCATTAATTCGGGATATACGTAGGAGGGGTAAGAATAAAGTGGCTGCTCAGAATTGCAGAAAAAGAAAACTGGAAAATATAGTAGAACTAGAGCAAGATTTAGATCATTTGAAAGATGAAAAAGAAAAATTGCTCAAAGAAAAAGGAGAAAATGACAAAAGCCTTCACCTACTGAAAAAACAACTCAGCACC"
#> NFE2L3
#> "GTCTCACTTATCCGTGACATCAGACGAAGAGGGAAAAATAAAGTTGCTGCGCAGAACTGTCGTAAACGCAAATTGGACATAATTTTGAATTTAGAAGATGATGTATGTAACTTGCAAGCAAAGAAGGAAACTCTTAAGAGAGAGCAAGCACAATGTAACAAAGCTATTAACATAATGAAACAGAAACTGCATGAC"
#> TEF
#> "CAGAAGGATGAAAAGTACTGGACAAGACGCAAGAAGAACAACGTGGCAGCTAAACGGTCACGGGATGCCCGGCGCCTGAAAGAGAATCAGATCACCATCCGGGCAGCCTTCCTGGAGAAGGAGAACACAGCCCTGCGGACGGAGGTGGCCGAGCTACGCAAGGAGGTGGGCAAGTGCAAGACCATCGTGTCCAAG"
#>
#> $wildTypeReverse
#> JUN
#> "ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT"
#>
#> $constantForward
#> [1] ""
#>
#> $constantReverse
#> [1] ""
#>
#> $avePhredMinForward
#> [1] 20
#>
#> $avePhredMinReverse
#> [1] 20
#>
#> $variableNMaxForward
#> [1] 0
#>
#> $variableNMaxReverse
#> [1] 0
#>
#> $umiNMax
#> [1] 0
#>
#> $nbrMutatedCodonsMaxForward
#> [1] 0
#>
#> $nbrMutatedCodonsMaxReverse
#> [1] 1
#>
#> $nbrMutatedBasesMaxForward
#> [1] -1
#>
#> $nbrMutatedBasesMaxReverse
#> [1] -1
#>
#> $forbiddenMutatedCodonsForward
#> character(0)
#>
#> $forbiddenMutatedCodonsReverse
#> [1] "AAA" "AAT" "ACA" "ACT" "AGA" "AGT" "ATA" "ATT" "CAA" "CAT" "CCA" "CCT"
#> [13] "CGA" "CGT" "CTA" "CTT" "GAA" "GAT" "GCA" "GCT" "GGA" "GGT" "GTA" "GTT"
#> [25] "TAA" "TAT" "TCA" "TCT" "TGA" "TGT" "TTA" "TTT"
#>
#> $useTreeWTmatch
#> [1] FALSE
#>
#> $collapseToWTForward
#> [1] FALSE
#>
#> $collapseToWTReverse
#> [1] FALSE
#>
#> $mutatedPhredMinForward
#> [1] 0
#>
#> $mutatedPhredMinReverse
#> [1] 0
#>
#> $mutNameDelimiter
#> [1] "."
#>
#> $constantMaxDistForward
#> [1] -1
#>
#> $constantMaxDistReverse
#> [1] -1
#>
#> $umiCollapseMaxDist
#> [1] 0
#>
#> $filteredReadsFastqForward
#> [1] ""
#>
#> $filteredReadsFastqReverse
#> [1] ""
#>
#> $maxNReads
#> [1] -1
#>
#> $nThreads
#> [1] 1
#>
#> $chunkSize
#> [1] 100000
#>
#> $maxReadLength
#> [1] 1024
#>
#> $processingInfo
#> [1] "Processed by mutscan v1.3.0 on 2026-04-28 18:01:11.381144"
leujunt0$filterSummary
#> nbrTotal f1_nbrAdapter f2_nbrNoPrimer f3_nbrReadWrongLength
#> 1 1000 0 126 0
#> f4_nbrNoValidOverlap f5_nbrAvgVarQualTooLow f6_nbrTooManyNinVar
#> 1 0 0 76
#> f7_nbrTooManyNinUMI f8_nbrTooManyBestWTHits f9_nbrMutQualTooLow
#> 1 0 0 0
#> f10a_nbrTooManyMutCodons f10b_nbrTooManyMutBases f11_nbrForbiddenCodons
#> 1 195 0 3
#> f12_nbrTooManyMutConstant f13_nbrTooManyBestConstantHits nbrRetained
#> 1 0 0 600
head(leujunt0$summaryTable)
#> mutantName
#> 1 ATF2.0.WT_JUN.13.CCC
#> 2 ATF2.0.WT_JUN.20.TGG
#> 3 ATF2.0.WT_JUN.32.CGG
#> 4 ATF2.0.WT_JUN.4.ATG
#> 5 ATF2.0.WT_JUN.5.ATG
#> 6 ATF2.0.WT_JUN.6.GAG
#> sequence
#> 1 GATCCTGATGAAAAAAGGAGAAAGTTTTTAGAGCGAAATAGAGCAGCAGCTTCAAGATGCCGACAAAAAAGGAAAGTCTGGGTTCAGTCTTTAGAGAAGAAAGCT_ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAACCCCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> 2 GATCCTGATGAAAAAAGGAGAAAGTTTTTAGAGCGAAATAGAGCAGCAGCTTCAAGATGCCGACAAAAAAGGAAAGTCTGGGTTCAGTCTTTAGAGAAGAAAG_ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTGGACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> 3 GATCCTGATGAAAAAAGGAGAAAGTTTTTAGAGCGAAATAGAGCAGCAGCTTCAAGATGCCGACAAAAAAGGAAAGTCTGGGTTCAGTCTTTAGAGAAGAAAGC_ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCGG
#> 4 GATCCTGATGAAAAAAGGAGAAAGTTTTTAGAGCGAAATAGAGCAGCAGCTTCAAGATGCCGACAAAAAAGGAAAGTCTGGGTTCAGTCTTTAGAGAAGAAAGC_ATCGCCCGGATGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> 5 GATCCTGATGAAAAAAGGAGAAAGTTTTTAGAGCGAAATAGAGCAGCAGCTTCAAGATGCCGACAAAAAAGGAAAGTCTGGGTTCAGTCTTTAGAGAAGAAAGCT_ATCGCCCGGCTGATGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> 6 GATCCTGATGAAAAAAGGAGAAAGTTTTTAGAGCGAAATAGAGCAGCAGCTTCAAGATGCCGACAAAAAAGGAAAGTCTGGGTTCAGTCTTTAGAGAAGAAAGCT_ATCGCCCGGCTGGAGGAGAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT
#> nbrReads maxNbrReads nbrUmis nbrMutBases nbrMutCodons nbrMutAAs varLengths
#> 1 1 1 0 2 1 1 105_96
#> 2 1 1 0 2 1 1 103_96
#> 3 1 1 0 2 1 1 104_96
#> 4 1 1 0 1 1 1 104_96
#> 5 1 1 0 2 1 1 105_96
#> 6 1 1 0 1 1 0 105_96
#> mutantNameBase mutantNameCodon mutantNameBaseHGVS
#> 1 ATF2.0.WT_JUN.37.C_JUN.39.C ATF2.0.WT_JUN.13.CCC ATF2:c_JUN:c.37_39delinsCCC
#> 2 ATF2.0.WT_JUN.59.G_JUN.60.G ATF2.0.WT_JUN.20.TGG ATF2:c_JUN:c.59_60delinsGG
#> 3 ATF2.0.WT_JUN.95.G_JUN.96.G ATF2.0.WT_JUN.32.CGG ATF2:c_JUN:c.95_96delinsGG
#> 4 ATF2.0.WT_JUN.10.A ATF2.0.WT_JUN.4.ATG ATF2:c_JUN:c.10C>A
#> 5 ATF2.0.WT_JUN.13.A_JUN.14.T ATF2.0.WT_JUN.5.ATG ATF2:c_JUN:c.13_14delinsAT
#> 6 ATF2.0.WT_JUN.18.G ATF2.0.WT_JUN.6.GAG ATF2:c_JUN:c.18A>G
#> mutantNameAA mutantNameAAHGVS mutationTypes
#> 1 ATF2.0.WT_JUN.13.P ATF2:p_JUN:p.(Ala13Pro) nonsynonymous
#> 2 ATF2.0.WT_JUN.20.W ATF2:p_JUN:p.(Ser20Trp) nonsynonymous
#> 3 ATF2.0.WT_JUN.32.R ATF2:p_JUN:p.(Leu32Arg) nonsynonymous
#> 4 ATF2.0.WT_JUN.4.M ATF2:p_JUN:p.(Leu4Met) nonsynonymous
#> 5 ATF2.0.WT_JUN.5.M ATF2:p_JUN:p.(Glu5Met) nonsynonymous
#> 6 ATF2.0.WT_JUN.0.WT ATF2:p_JUN:p silent
#> sequenceAA
#> 1 DPDEKRRKFLERNRAAASRCRQKRKVWVQSLEKKA_IARLEEKVKTLKPQNSELASTANMLREQVAQL
#> 2 DPDEKRRKFLERNRAAASRCRQKRKVWVQSLEKK_VARLEEKVKTLKAQNSELAWTANMLREQVAQL
#> 3 DPDEKRRKFLERNRAAASRCRQKRKVWVQSLEKK_EARLEEKVKTLKAQNSELASTANMLREQVAQR
#> 4 DPDEKRRKFLERNRAAASRCRQKRKVWVQSLEKK_EARMEEKVKTLKAQNSELASTANMLREQVAQL
#> 5 DPDEKRRKFLERNRAAASRCRQKRKVWVQSLEKKA_IARLMEKVKTLKAQNSELASTANMLREQVAQL
#> 6 DPDEKRRKFLERNRAAASRCRQKRKVWVQSLEKKA_IARLEEKVKTLKAQNSELASTANMLREQVAQLCombining samples
The digestFastqs() function processes each sample (pair
of FASTQ files) separately. In order to prepare the data for downstream
statistical analysis and plotting, the
summarizeExperiment() function takes a named list of
outputs from digestFastqs(), and a data.frame
with sample annotations, and generates a
SummarizedExperiment object, with an assay
containing either UMI or read counts. To illustrate this, we process
also the output sample for the TRANS experiment for which we processed
the input sample above, and feed both outputs to
summarizeExperiment().
transOutput <- digestFastqs(
fastqForward = file.path(datadir, "transOutput_1.fastq.gz"),
fastqReverse = file.path(datadir, "transOutput_2.fastq.gz"),
mergeForwardReverse = FALSE,
revComplForward = FALSE,
revComplReverse = FALSE,
adapterForward = "GGAAGAGCACACGTC",
adapterReverse = "GGAAGAGCGTCGTGT",
elementsForward = "SUCV",
elementsReverse = "SUCV",
elementLengthsForward = c(1, 10, 18, 96),
elementLengthsReverse = c(1, 8, 20, 96),
constantForward = "AACCGGAGGAGGGAGCTG",
constantReverse = "GAAAAAGGAAGCTGGAGAGA",
primerForward = "",
primerReverse = "",
avePhredMinForward = 20,
avePhredMinReverse = 20,
variableNMaxForward = 0,
variableNMaxReverse = 0,
umiNMax = 0,
wildTypeForward = c(FOS = "ACTGATACACTCCAAGCGGAGACAGACCAACTAGAAGATGAGAAGTCTGCTTTGCAGACCGAGATTGCCAACCTGCTGAAGGAGAAGGAAAAACTA"),
wildTypeReverse = c(JUN = "ATCGCCCGGCTGGAGGAAAAAGTGAAAACCTTGAAAGCTCAGAACTCGGAGCTGGCGTCCACGGCCAACATGCTCAGGGAACAGGTGGCACAGCTT"),
nbrMutatedCodonsMaxForward = 1,
nbrMutatedCodonsMaxReverse = 1,
forbiddenMutatedCodonsForward = "NNW",
forbiddenMutatedCodonsReverse = "NNW",
mutNameDelimiter = ".",
constantMaxDistForward = -1,
constantMaxDistReverse = -1,
verbose = FALSE
)
## Generate SummarizedExperiment object
se <- summarizeExperiment(
x = list(sample1 = transInput,
sample2 = transOutput),
coldata = data.frame(Name = c("sample1", "sample2"),
Condition = c("input", "output"),
Replicate = c("R1", "R1"),
OD = c(0.05, 1.5))
)
## The SummarizedExperiment contains a count matrix, and annotations
## for samples and variants in the `colData` and `rowData`,
## respectively.
head(assay(se, "counts"))
#> 6 x 2 sparse Matrix of class "dgCMatrix"
#> sample1 sample2
#> FOS.0.WT_JUN.0.WT . 1
#> FOS.0.WT_JUN.13.CCC 1 .
#> FOS.0.WT_JUN.13.CTC 1 .
#> FOS.0.WT_JUN.13.GCG . 1
#> FOS.0.WT_JUN.2.TCC 1 .
#> FOS.0.WT_JUN.20.ACC 1 .
Matrix::colSums(assay(se, "counts"))
#> sample1 sample2
#> 279 285
head(rowData(se))
#> DataFrame with 6 rows and 19 columns
#> mutantName sequence nbrMutBases
#> <character> <character> <character>
#> FOS.0.WT_JUN.0.WT FOS.0.WT_JUN.0.WT ACTGATACACTCCAAGCGGA.. 0
#> FOS.0.WT_JUN.13.CCC FOS.0.WT_JUN.13.CCC ACTGATACACTCCAAGCGGA.. 2
#> FOS.0.WT_JUN.13.CTC FOS.0.WT_JUN.13.CTC ACTGATACACTCCAAGCGGA.. 3
#> FOS.0.WT_JUN.13.GCG FOS.0.WT_JUN.13.GCG ACTGATACACTCCAAGCGGA.. 1
#> FOS.0.WT_JUN.2.TCC FOS.0.WT_JUN.2.TCC ACTGATACACTCCAAGCGGA.. 1
#> FOS.0.WT_JUN.20.ACC FOS.0.WT_JUN.20.ACC ACTGATACACTCCAAGCGGA.. 1
#> minNbrMutBases maxNbrMutBases nbrMutCodons minNbrMutCodons
#> <integer> <integer> <character> <integer>
#> FOS.0.WT_JUN.0.WT 0 0 0 0
#> FOS.0.WT_JUN.13.CCC 2 2 1 1
#> FOS.0.WT_JUN.13.CTC 3 3 1 1
#> FOS.0.WT_JUN.13.GCG 1 1 1 1
#> FOS.0.WT_JUN.2.TCC 1 1 1 1
#> FOS.0.WT_JUN.20.ACC 1 1 1 1
#> maxNbrMutCodons nbrMutAAs minNbrMutAAs maxNbrMutAAs
#> <integer> <character> <integer> <integer>
#> FOS.0.WT_JUN.0.WT 0 0 0 0
#> FOS.0.WT_JUN.13.CCC 1 1 1 1
#> FOS.0.WT_JUN.13.CTC 1 1 1 1
#> FOS.0.WT_JUN.13.GCG 1 0 0 0
#> FOS.0.WT_JUN.2.TCC 1 1 1 1
#> FOS.0.WT_JUN.20.ACC 1 1 1 1
#> mutantNameBase mutantNameBaseHGVS
#> <character> <character>
#> FOS.0.WT_JUN.0.WT FOS.0.WT_JUN.0.WT FOS:c_JUN:c
#> FOS.0.WT_JUN.13.CCC FOS.0.WT_JUN.37.C_JU.. FOS:c_JUN:c.37_39del..
#> FOS.0.WT_JUN.13.CTC FOS.0.WT_JUN.37.C_JU.. FOS:c_JUN:c.37_39del..
#> FOS.0.WT_JUN.13.GCG FOS.0.WT_JUN.39.G FOS:c_JUN:c.39T>G
#> FOS.0.WT_JUN.2.TCC FOS.0.WT_JUN.4.T FOS:c_JUN:c.4G>T
#> FOS.0.WT_JUN.20.ACC FOS.0.WT_JUN.58.A FOS:c_JUN:c.58T>A
#> mutantNameCodon mutantNameAA
#> <character> <character>
#> FOS.0.WT_JUN.0.WT FOS.0.WT_JUN.0.WT FOS.0.WT_JUN.0.WT
#> FOS.0.WT_JUN.13.CCC FOS.0.WT_JUN.13.CCC FOS.0.WT_JUN.13.P
#> FOS.0.WT_JUN.13.CTC FOS.0.WT_JUN.13.CTC FOS.0.WT_JUN.13.L
#> FOS.0.WT_JUN.13.GCG FOS.0.WT_JUN.13.GCG FOS.0.WT_JUN.0.WT
#> FOS.0.WT_JUN.2.TCC FOS.0.WT_JUN.2.TCC FOS.0.WT_JUN.2.S
#> FOS.0.WT_JUN.20.ACC FOS.0.WT_JUN.20.ACC FOS.0.WT_JUN.20.T
#> mutantNameAAHGVS sequenceAA mutationTypes
#> <character> <character> <character>
#> FOS.0.WT_JUN.0.WT FOS:p_JUN:p TDTLQAETDQLEDEKSALQT..
#> FOS.0.WT_JUN.13.CCC FOS:p_JUN:p.(Ala13Pro) TDTLQAETDQLEDEKSALQT.. nonsynonymous
#> FOS.0.WT_JUN.13.CTC FOS:p_JUN:p.(Ala13Leu) TDTLQAETDQLEDEKSALQT.. nonsynonymous
#> FOS.0.WT_JUN.13.GCG FOS:p_JUN:p TDTLQAETDQLEDEKSALQT.. silent
#> FOS.0.WT_JUN.2.TCC FOS:p_JUN:p.(Ala2Ser) TDTLQAETDQLEDEKSALQT.. nonsynonymous
#> FOS.0.WT_JUN.20.ACC FOS:p_JUN:p.(Ser20Thr) TDTLQAETDQLEDEKSALQT.. nonsynonymous
#> varLengths
#> <character>
#> FOS.0.WT_JUN.0.WT 96_96
#> FOS.0.WT_JUN.13.CCC 96_96
#> FOS.0.WT_JUN.13.CTC 96_96
#> FOS.0.WT_JUN.13.GCG 96_96
#> FOS.0.WT_JUN.2.TCC 96_96
#> FOS.0.WT_JUN.20.ACC 96_96
colData(se)
#> DataFrame with 2 rows and 20 columns
#> Name Condition Replicate OD nbrTotal f1_nbrAdapter
#> <character> <character> <character> <numeric> <integer> <integer>
#> sample1 sample1 input R1 0.05 1000 314
#> sample2 sample2 output R1 1.50 1000 366
#> f2_nbrNoPrimer f3_nbrReadWrongLength f4_nbrNoValidOverlap
#> <integer> <integer> <integer>
#> sample1 0 0 0
#> sample2 0 0 0
#> f5_nbrAvgVarQualTooLow f6_nbrTooManyNinVar f7_nbrTooManyNinUMI
#> <integer> <integer> <integer>
#> sample1 7 0 0
#> sample2 6 0 0
#> f8_nbrTooManyBestWTHits f9_nbrMutQualTooLow f10a_nbrTooManyMutCodons
#> <integer> <integer> <integer>
#> sample1 0 0 392
#> sample2 0 0 338
#> f10b_nbrTooManyMutBases f11_nbrForbiddenCodons
#> <integer> <integer>
#> sample1 0 8
#> sample2 0 5
#> f12_nbrTooManyMutConstant f13_nbrTooManyBestConstantHits nbrRetained
#> <integer> <integer> <integer>
#> sample1 0 0 279
#> sample2 0 0 285
## Count type (reads or UMIs)
metadata(se)$countType
#> [1] "umis"Collapsing count matrix to amino acids
The object generated by summarizeExperiment() contains
one row for each observed variant (combination). This can be further
collapsed by replacing the mutated codon by the corresponding amino
acid, and aggregating the counts corresponding to the same mutated amino
acid (combination).
se_collapsed <- collapseMutantsByAA(se)
head(assay(se_collapsed, "counts"))
#> sample1 sample2
#> FOS.0.WT_JUN.0.WT 1 3
#> FOS.0.WT_JUN.10.S 0 1
#> FOS.0.WT_JUN.12.L 0 1
#> FOS.0.WT_JUN.13.K 1 0
#> FOS.0.WT_JUN.13.L 1 0
#> FOS.0.WT_JUN.13.P 1 0
Matrix::colSums(assay(se_collapsed, "counts"))
#> sample1 sample2
#> 279 285
colData(se_collapsed)
#> DataFrame with 2 rows and 20 columns
#> Name Condition Replicate OD nbrTotal f1_nbrAdapter
#> <character> <character> <character> <numeric> <integer> <integer>
#> sample1 sample1 input R1 0.05 1000 314
#> sample2 sample2 output R1 1.50 1000 366
#> f2_nbrNoPrimer f3_nbrReadWrongLength f4_nbrNoValidOverlap
#> <integer> <integer> <integer>
#> sample1 0 0 0
#> sample2 0 0 0
#> f5_nbrAvgVarQualTooLow f6_nbrTooManyNinVar f7_nbrTooManyNinUMI
#> <integer> <integer> <integer>
#> sample1 7 0 0
#> sample2 6 0 0
#> f8_nbrTooManyBestWTHits f9_nbrMutQualTooLow f10a_nbrTooManyMutCodons
#> <integer> <integer> <integer>
#> sample1 0 0 392
#> sample2 0 0 338
#> f10b_nbrTooManyMutBases f11_nbrForbiddenCodons
#> <integer> <integer>
#> sample1 0 8
#> sample2 0 5
#> f12_nbrTooManyMutConstant f13_nbrTooManyBestConstantHits nbrRetained
#> <integer> <integer> <integer>
#> sample1 0 0 279
#> sample2 0 0 285Diagnostic plots
mutscan contains functionality for generating a variety
of diagnostic plots. Here we illustrate these using the full CIS data
set from [@Diss2018], which has been
processed using digestFastqs() as illustrated above, and
summarized in a SummarizedExperiment object provided with
the package.
First, we can plot a summary of the filtering process, indicating the
number of reads that were filtered out by (or retained after) each step
of the mutscan filtering.
plotFiltering(se, valueType = "reads", onlyActiveFilters = TRUE,
plotType = "remaining", facetBy = "sample", numberSize = 3)
plotFiltering(se, valueType = "fractions", onlyActiveFilters = TRUE,
plotType = "filtered", facetBy = "step", numberSize = 3)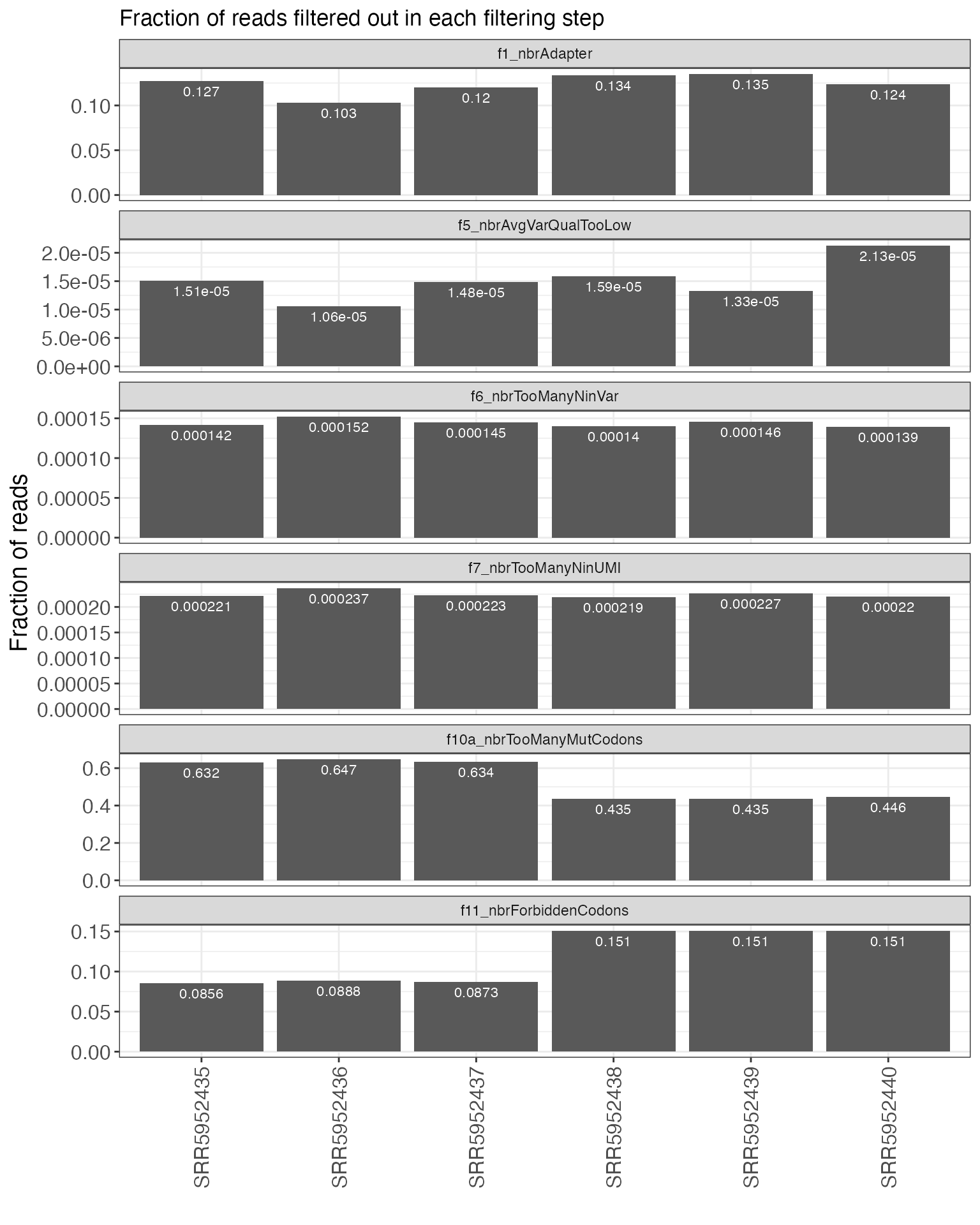
We can also generate a pairs plot displaying the correlation among the samples in the data set.
plotPairs(se, selAssay = "counts")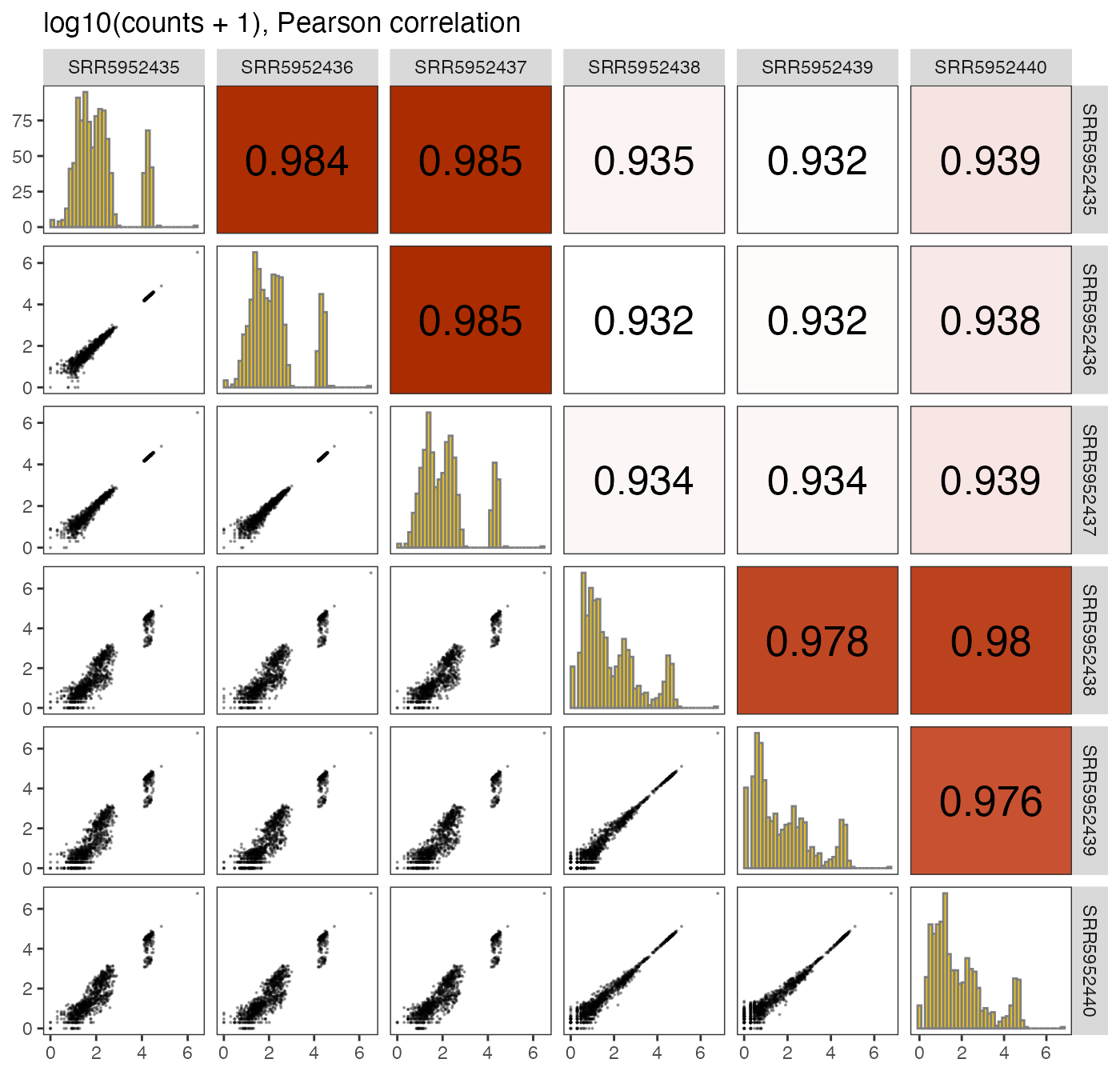
Additional plot functions can be used to visualize the total count per sample, across all variants, or the distribution of variant counts per sample.
plotTotals(se, selAssay = "counts")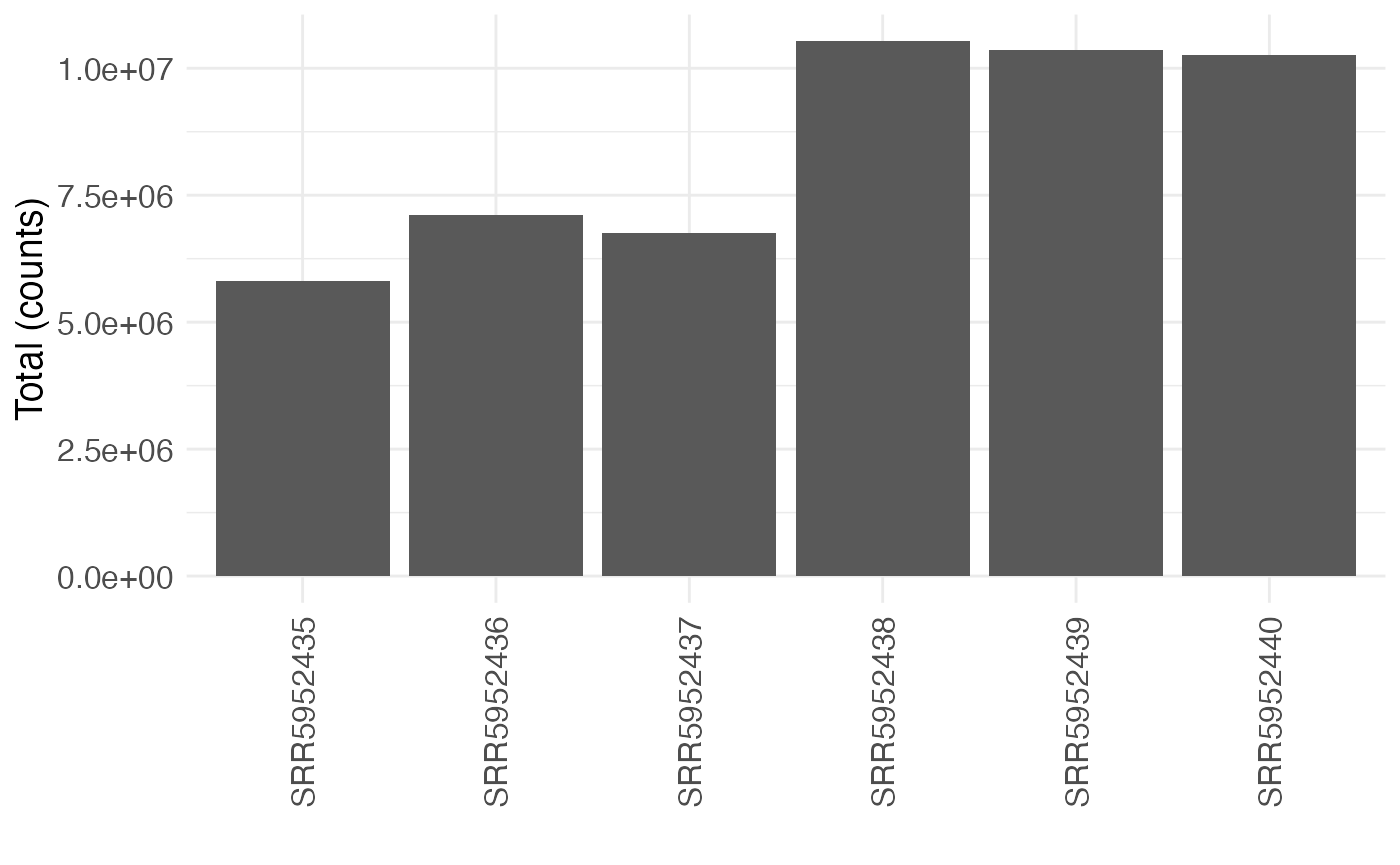
plotDistributions(se, selAssay = "counts", plotType = "density",
pseudocount = 1)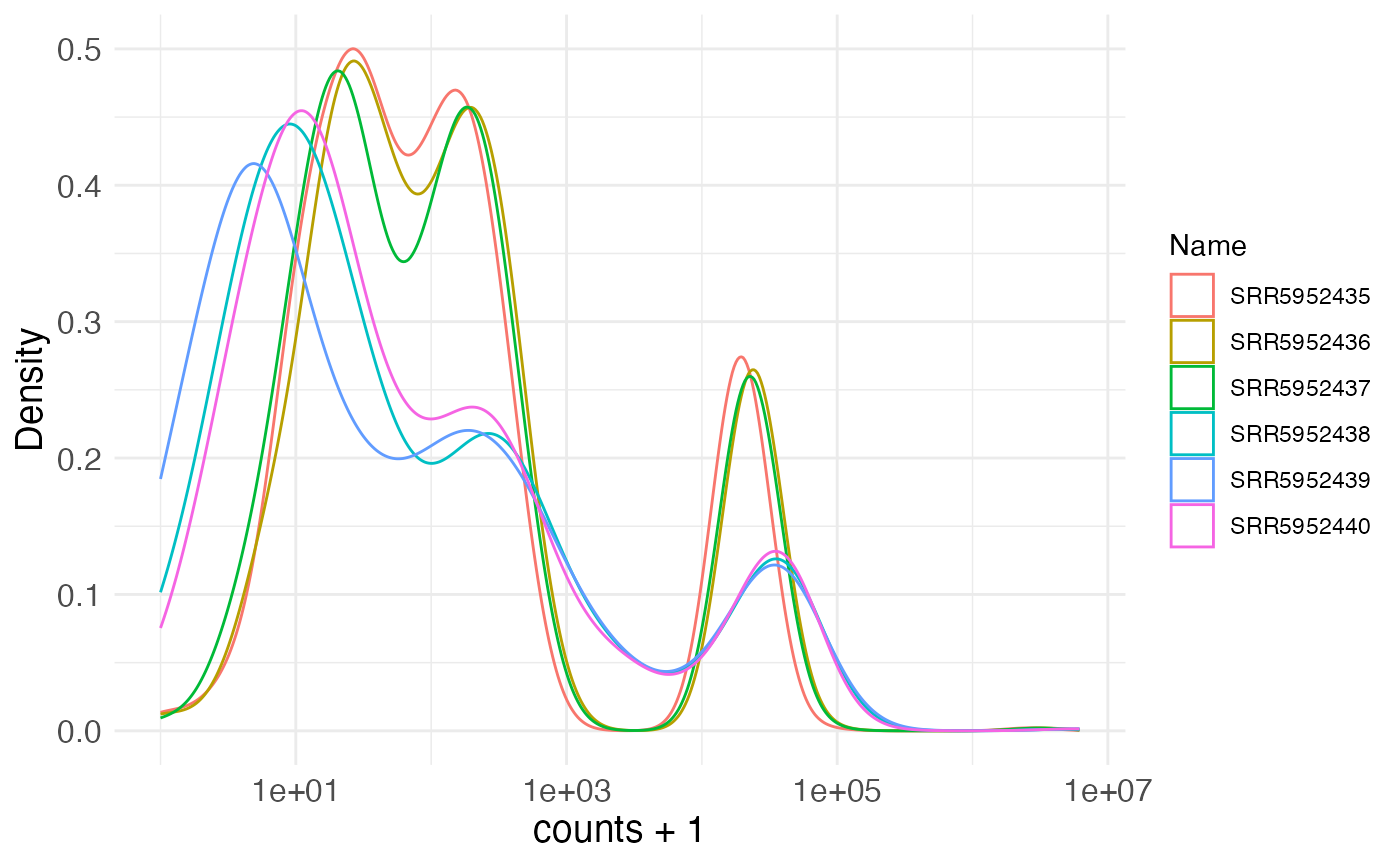
Finally, we can create a full QC report as follows:
generateQCReport(se, outFile = tempfile(fileext = ".html"))Calculating fitness scores
The function calculateFitnessScore() can be used to
calculate fitness scores as described in [@Diss2018]. The function requires the user to
specify a pairingCol, containing the replicate ID for each
sample; one or more ODCols, containing the optical density
for each sample, and a comparison, which is a character
vector of length 3 specifying the comparison to perform, of the form
(groupColumn, numerator,
denominator). Here, groupColumn is the name of
the column in colData(se) that contains the grouping
information, and numerator and denominator
specify the values of this column representing the two groups to be
compared.
Here, we illustrate the application of
calculateFitnessScore() on the SummarizedExperiment object
containing all the three CIS replicates from [@Diss2018].
se_collapsed <- collapseMutantsByAA(se)
ppis <- calculateFitnessScore(
se = se_collapsed, pairingCol = "Replicate",
ODCols = c("OD1", "OD2"),
comparison = c("Condition", "cis_output", "cis_input"),
WTrows = "f.0.WT"
)
head(ppis[order(abs(rowMeans(ppis)), decreasing = TRUE), , drop = FALSE])
#> cis_output_vs_cis_input_repl1 cis_output_vs_cis_input_repl2
#> f.8.M 1.079682 1.077555
#> f.8.A 1.077780 1.087804
#> f.2.Q 1.093753 1.065724
#> f.3.M 1.084323 1.078138
#> f.9.K 1.085896 1.037838
#> f.13.K 1.043217 1.046260
#> cis_output_vs_cis_input_repl3
#> f.8.M 1.117395
#> f.8.A 1.103149
#> f.2.Q 1.065259
#> f.3.M 1.050588
#> f.9.K 1.062279
#> f.13.K 1.095404
## The PPI score for the WT sequence is 1, by construction
ppis["f.0.WT", , drop = FALSE]
#> cis_output_vs_cis_input_repl1 cis_output_vs_cis_input_repl2
#> f.0.WT 1 1
#> cis_output_vs_cis_input_repl3
#> f.0.WT 1Scoring mutations with edgeR or limma
As an alternative to the fitness scoring, mutscan can be
used to model the observed counts using a generalized linear model (with
edgeR) or a general linear model (with limma)
and estimate a logFC and a p-value for the enrichment of each variant
betwen two conditions (or more generally, in association with any
predictor), compared to one or more WT sequences. Note that for this, at
least two replicates are required.
We start by looking at the design matrix, in order to determine which
of the coefficients to specify for the testing in
calculateRelativeFC(). For more information about how to
set up and interpret design matrices in edgeR or
limma, see e.g. Law et al (2020),
or Soneson et al
(2020).
model.matrix(~ Replicate + Condition,
data = colData(se_collapsed))
#> (Intercept) Replicate Conditioncis_output
#> SRR5952435 1 1 0
#> SRR5952436 1 2 0
#> SRR5952437 1 3 0
#> SRR5952438 1 1 1
#> SRR5952439 1 2 1
#> SRR5952440 1 3 1
#> attr(,"assign")
#> [1] 0 1 2
#> attr(,"contrasts")
#> attr(,"contrasts")$Condition
#> [1] "contr.treatment"Next, we apply either edgeR or limma to
extract the logFCs of the mutants, compared to the wildtype
sequence.
## edgeR
edger_scores <- calculateRelativeFC(
se = se_collapsed,
design = model.matrix(~ Replicate + Condition,
data = colData(se_collapsed)),
coef = "Conditioncis_output", pseudocount = 1, WTrows = "f.0.WT",
method = "edgeR")
head(edger_scores[order(edger_scores$PValue), , drop = FALSE])
#> logFC logCPM F PValue FDR logFC_shrunk
#> f.27.* -4.503507 11.12474 14566.17 2.086636e-24 9.061367e-22 -4.502824
#> f.7.G -4.184495 11.14499 13607.70 3.528342e-24 9.061367e-22 -4.183994
#> f.28.* -4.387507 11.14116 13284.48 4.247516e-24 9.061367e-22 -4.386944
#> f.7.* -4.460185 10.87738 12511.63 6.745260e-24 9.663684e-22 -4.459450
#> f.14.* -4.373711 10.90447 12285.63 7.763935e-24 9.663684e-22 -4.372984
#> f.28.G -3.942580 11.21716 11850.16 1.025681e-23 9.663684e-22 -3.942115
#> df.total df.prior df.test
#> f.27.* 15.45022 12.45019 1
#> f.7.G 15.45022 12.45019 1
#> f.28.* 15.45022 12.45019 1
#> f.7.* 15.45022 12.45019 1
#> f.14.* 15.45022 12.45019 1
#> f.28.G 15.45022 12.45019 1
## As before, the WT sequence has a logFC close to 0, by construction
edger_scores["f.0.WT", , drop = FALSE]
#> logFC logCPM F PValue FDR logFC_shrunk df.total df.prior
#> f.0.WT 9.039516e-16 19.30433 0 1 1 -2.364216e-15 15.45022 12.45019
#> df.test
#> f.0.WT 1
## limma
limma_scores <- calculateRelativeFC(
se = se_collapsed,
design = model.matrix(~ Replicate + Condition,
data = colData(se_collapsed)),
coef = "Conditioncis_output", pseudocount = 1, WTrows = "f.0.WT",
method = "limma")
head(limma_scores[order(limma_scores$P.Value), , drop = FALSE])
#> logFC CI.L CI.R AveExpr t P.Value
#> f.27.* -4.503210 -4.622021 -4.384400 9.963039 -84.07624 5.299650e-16
#> f.4.F -2.897232 -2.974240 -2.820225 11.116524 -83.45582 5.720907e-16
#> f.28.* -4.387941 -4.505027 -4.270855 10.031042 -83.13096 5.956021e-16
#> f.7.G -4.183176 -4.296040 -4.070312 10.124077 -82.21606 6.677075e-16
#> f.21.G -4.080241 -4.196485 -3.963998 10.116854 -77.86148 1.171161e-15
#> f.29.* -4.469138 -4.596492 -4.341784 10.036584 -77.84277 1.174071e-15
#> adj.P.Val B se.logFC df.total df.prior
#> f.27.* 1.068332e-13 27.23208 0.05356103 10.34072 7.340722
#> f.4.F 1.068332e-13 27.37901 0.03471576 10.34072 7.340722
#> f.28.* 1.068332e-13 27.16244 0.05278347 10.34072 7.340722
#> f.7.G 1.068332e-13 27.10387 0.05088028 10.34072 7.340722
#> f.21.G 1.077888e-13 26.58967 0.05240385 10.34072 7.340722
#> f.29.* 1.077888e-13 26.53937 0.05741237 10.34072 7.340722
## As before, the WT sequence has a logFC close to 0, by construction
limma_scores["f.0.WT", , drop = FALSE]
#> logFC CI.L CI.R AveExpr t P.Value
#> f.0.WT 2.834536e-08 -0.02280783 0.02280789 19.29458 2.756789e-06 0.9999979
#> adj.P.Val B se.logFC df.total df.prior
#> f.0.WT 0.9999979 -10.20615 0.01028202 10.34072 7.340722mutscan also contains functions for plotting the results
of the statistical testing - in particular, MA (mean-difference) plots
and volcano plots can be easily generated.
plotMeanDiff(edger_scores, pointSize = "large")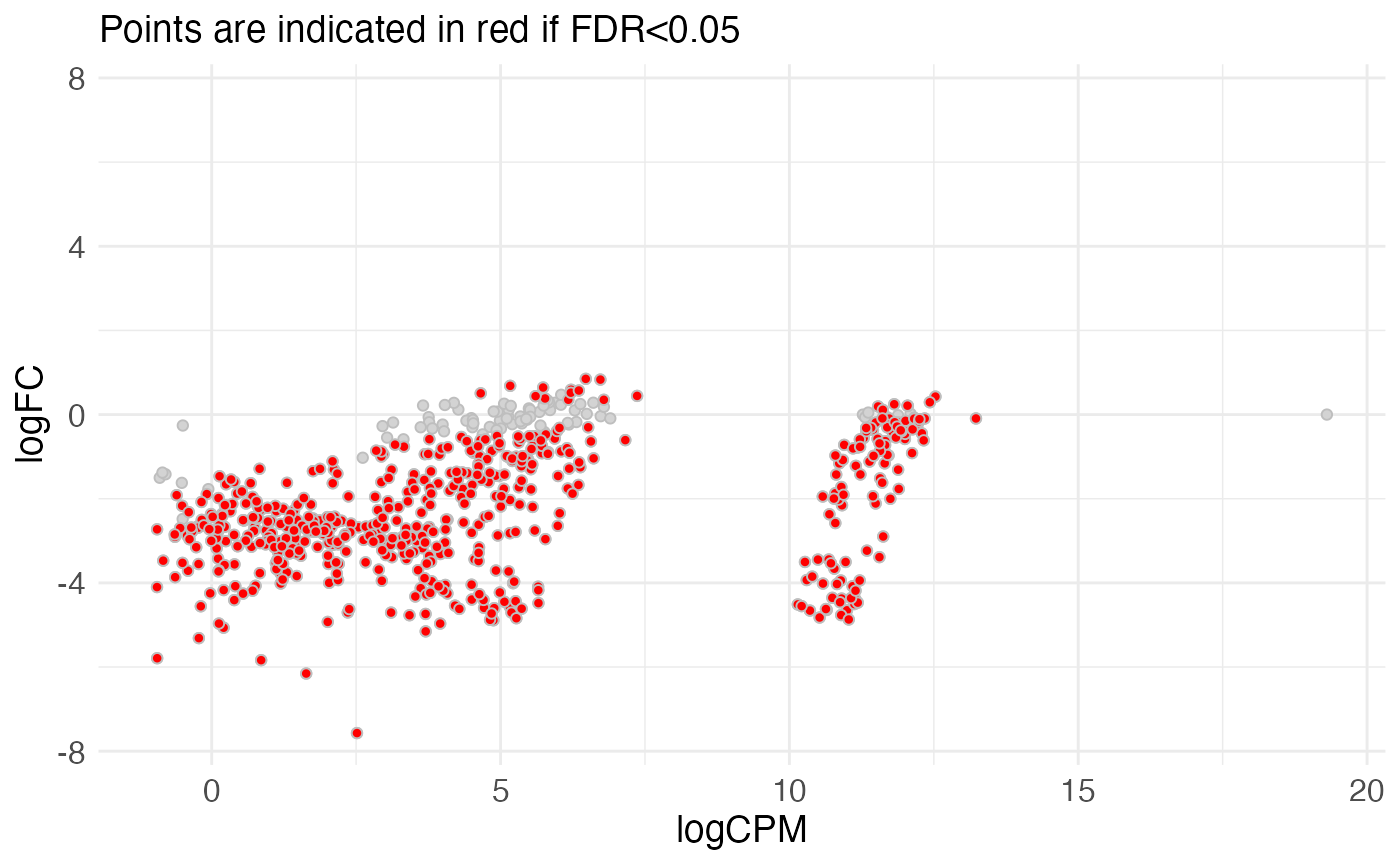
plotVolcano(edger_scores, pointSize = "large")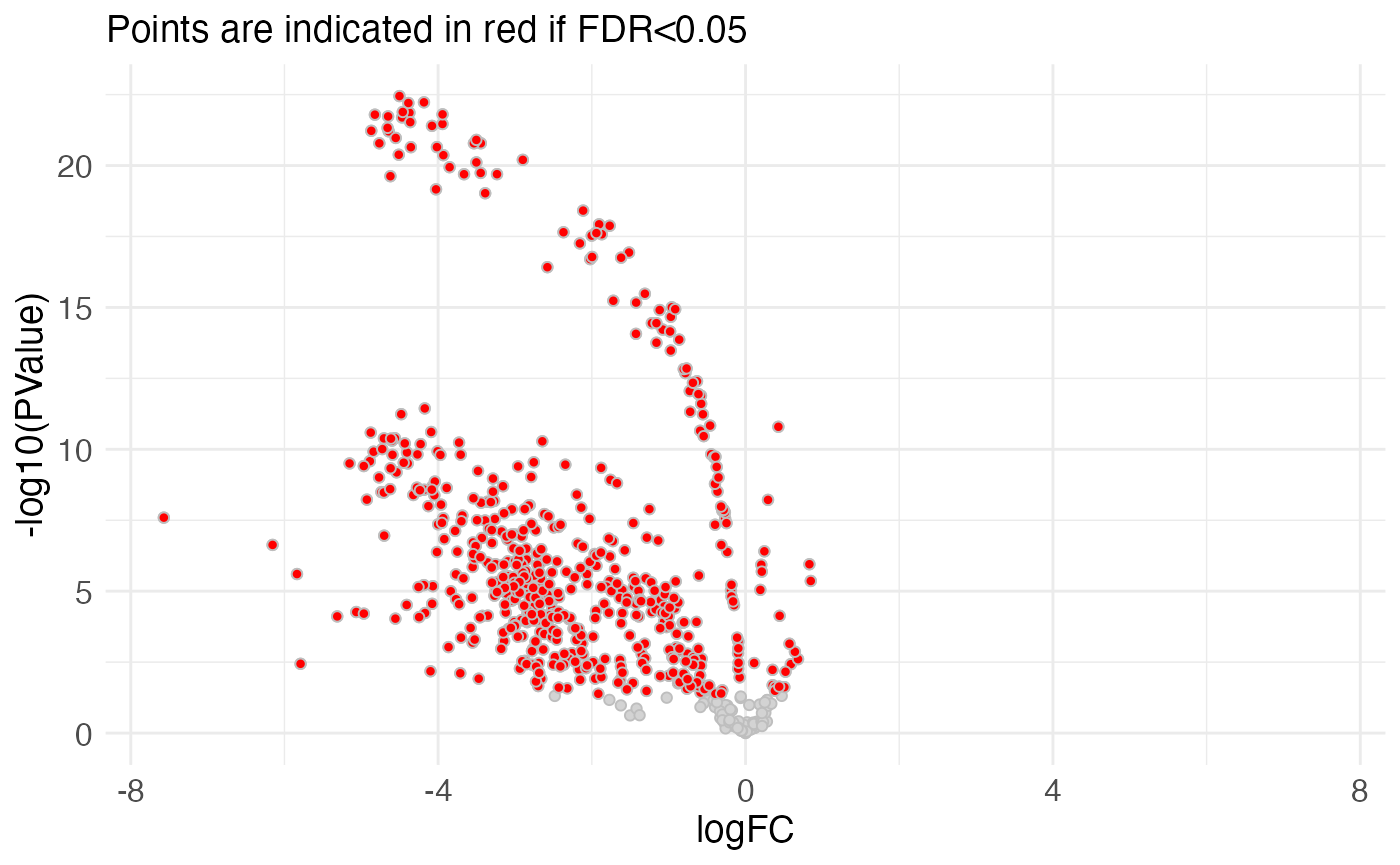
FAQ
Can digestFastqs process a sample where the reads are
spread across multiple (pairs of) FASTQ files?
Yes, you can specify a vector of FASTQ files to
fastqForward/fastqReverse. In this case, the
reads from all (pairs of) files will be analyzed as if they came from a
single FASTQ file. In case of paired-end data, take care to specify the
forward and reverse FASTQ files in the same order.
Session info
This vignette was compiled on the following system:
sessionInfo()
#> R version 4.6.0 Patched (2026-04-27 r89967)
#> Platform: aarch64-apple-darwin23
#> Running under: macOS Sequoia 15.7.4
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.6/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.6/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: UTC
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] mutscan_1.3.0 SummarizedExperiment_1.41.1
#> [3] Biobase_2.71.0 GenomicRanges_1.63.2
#> [5] Seqinfo_1.1.0 IRanges_2.45.0
#> [7] S4Vectors_0.49.2 BiocGenerics_0.57.1
#> [9] generics_0.1.4 MatrixGenerics_1.23.0
#> [11] matrixStats_1.5.0 BiocStyle_2.39.0
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 xfun_0.57 bslib_0.10.0
#> [4] ggplot2_4.0.3 htmlwidgets_1.6.4 ggrepel_0.9.8
#> [7] GGally_2.4.0 lattice_0.22-9 bitops_1.0-9
#> [10] vctrs_0.7.3 tools_4.6.0 parallel_4.6.0
#> [13] tibble_3.3.1 pkgconfig_2.0.3 Matrix_1.7-5
#> [16] RColorBrewer_1.1-3 S7_0.2.2 desc_1.4.3
#> [19] lifecycle_1.0.5 compiler_4.6.0 farver_2.1.2
#> [22] Biostrings_2.79.5 Rsamtools_2.27.2 textshaping_1.0.5
#> [25] statmod_1.5.1 codetools_0.2-20 htmltools_0.5.9
#> [28] sass_0.4.10 yaml_2.3.12 crayon_1.5.3
#> [31] pkgdown_2.2.0.9000 pillar_1.11.1 jquerylib_0.1.4
#> [34] tidyr_1.3.2 BiocParallel_1.45.0 DT_0.34.0
#> [37] limma_3.67.3 DelayedArray_0.37.1 cachem_1.1.0
#> [40] abind_1.4-8 ggstats_0.13.0 metapod_1.19.2
#> [43] locfit_1.5-9.12 tidyselect_1.2.1 digest_0.6.39
#> [46] dplyr_1.2.1 purrr_1.2.2 bookdown_0.46
#> [49] labeling_0.4.3 fastmap_1.2.0 grid_4.6.0
#> [52] cli_3.6.6 SparseArray_1.11.13 magrittr_2.0.5
#> [55] S4Arrays_1.11.1 withr_3.0.2 edgeR_4.9.9
#> [58] scales_1.4.0 rmarkdown_2.31 XVector_0.51.0
#> [61] otel_0.2.0 ragg_1.5.2 evaluate_1.0.5
#> [64] knitr_1.51 rlang_1.2.0 Rcpp_1.1.1-1.1
#> [67] glue_1.8.1 BiocManager_1.30.27 csaw_1.45.0
#> [70] jsonlite_2.0.0 R6_2.6.1 systemfonts_1.3.2
#> [73] fs_2.1.0