This chapter explains how to use SingleMoleculeGenomicsIO to filter single molecule footprinting data. The package contains functionality to calculate a collection of quality statistics for each read, and use these to filter out low-quality reads. This filtering can be done either on the imported SummarizedExperiment object (both for modBam and mismatchBam file types), or directly on the bam file (currently only modBam, which will generate another, filtered modBam file). SingleMoleculeGenomicsIO also provides several ways to filter out individual genomic positions based on, e.g. the genomic sequence or the read coverage.
We start by loading the required packages. In addition to the software package, we load a BSgenome object providing the mouse genome sequence.
To illustrate the position-level filtering of imported data, we first read 6mA data from a small genomic region for two samples. We add the sequence context (a single nucleotide) to be able to use this information as a basis for filtering.
Show/hide code
se <-readModBam(bamfiles =c(wt1 ="data/mESC_wt_6mA_rep1.bam",wt2 ="data/mESC_wt_6mA_rep2.bam"),modbase ="a", regions ="chr8:39286301-39287100", seqinfo =seqinfo(gnm), sequenceContextWidth =1, sequenceReference = gnm)se
The sequence information is stored in the rowData of se:
Show/hide code
rowData(se)
DataFrame with 31693 rows and 1 column
sequenceContext
<DNAStringSet>
1 A
2 A
3 G
4 A
5 A
... ...
31689 A
31690 A
31691 A
31692 A
31693 A
Position filtering can now be performed with the filterPositions function. The filters argument define which filters to apply, as well as the order (sequence context, coverage, removal of positions without non-NA values). Here, we retain only positions where the genome sequence is an A, and the coverage (the number of overlapping reads) is at least five. The assayNameCov argument indicates which assay will be used to define the coverage. If this is a read-level assay (like here), coverage will first be calculated using flattenReadLevelAssay. For more precise control, the summary assay can also be manually calculated and added to se beforehand, and specified in assayNameCov.
Show/hide code
sefilt <-filterPositions( se, filters =c("sequenceContext", "coverage", "all.na"),sequenceContext ="A",assayNameCov ="mod_prob",minCov =5)sefilt
In this case, the position filtering reduced the number of unique positions from 31693 to 16729 by 47.2%. However, the number of non-NA values in the matrix is only reduced from 333127 to 304984 (8.4%), confirming that the filtered-out positions are generally covered only by few reads.
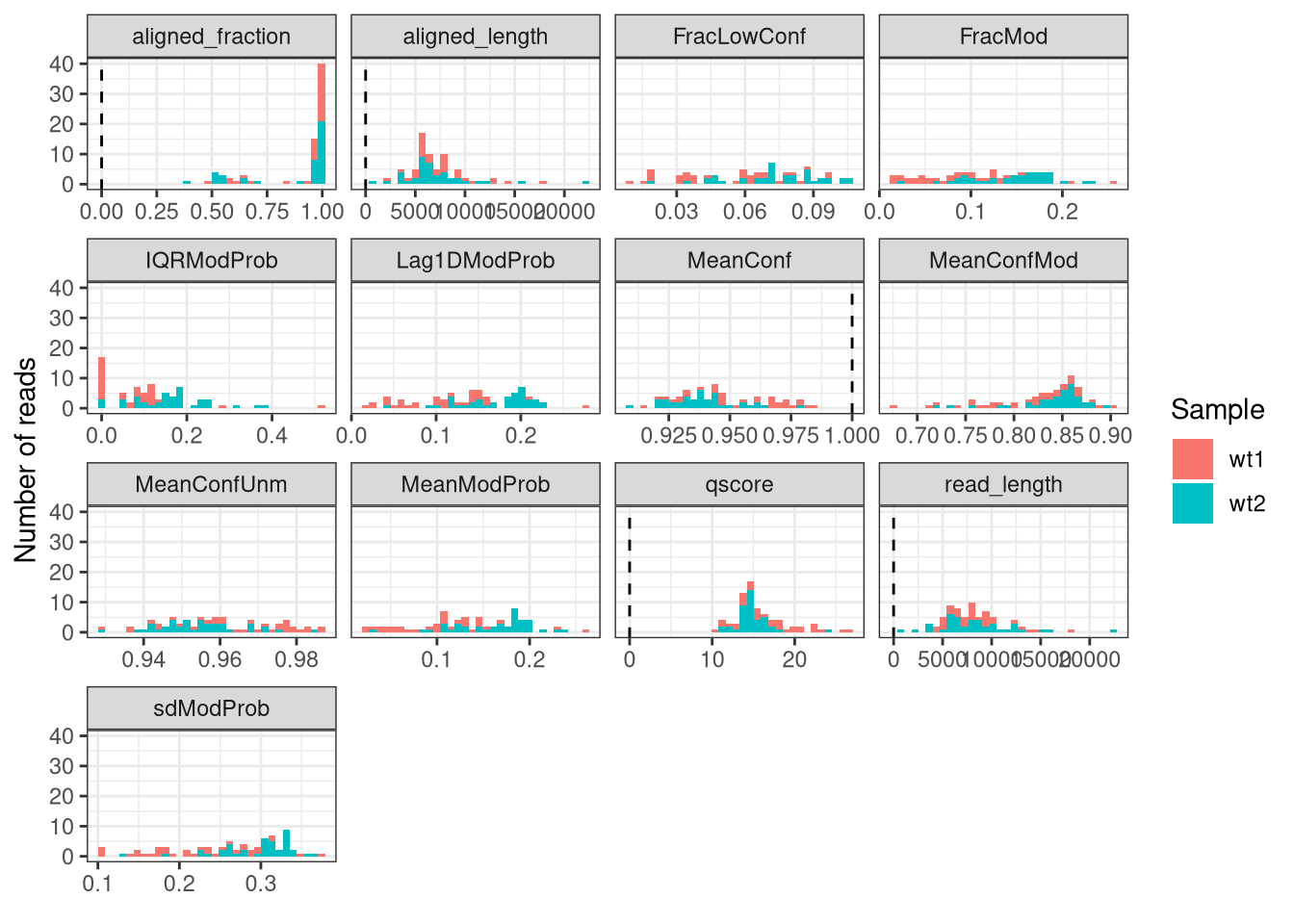
In addition to the position filtering illustrated above, SingleMoleculeGenomicsIO also provides functionality for calculating read-level quality scores and filtering out reads with low quality. The calculation of the quality scores is done using the addReadStats function, and the filtering is performed via the filterReads function.
Show/hide code
# Calculate read statisticssefilt <-addReadStats( sefilt, name ="QC")# The calculated read statistics are stored in the colDatacolData(sefilt)
DataFrame with 2 rows and 5 columns
sample modbase n_reads readInfo
<character> <character> <integer> <List>
wt1 wt1 a 41 14.1235:22453:22149:...,15.9814:15617:15460:...,14.3505:12359:12212:...,...
wt2 wt2 a 67 15.5769:39918:39406:...,15.4317:23376:23207:...,16.8065:23487:23313:...,...
QC
<List>
wt1 0.194044:0.186071:0.932636:...,0.157727:0.144965:0.947031:...,0.189361:0.176007:0.940345:...,...
wt2 0.217511:0.202377:0.941197:...,0.232775:0.221732:0.940212:...,0.161989:0.146135:0.947654:...,...
As mentioned above, SingleMoleculeGenomicsIO can also be used to directly filter alignments in a modBam file. The filterReadsModBam function reads the alignments in the input file, parses them, and writes them to the output file if they pass all the designated filters. These filters are treated hierarchically - in other words, if a read does not pass a given filter, the remaining filters will not be examined and the processing continues with the next read. Here we illustrate the modBam filtering using two small example modBam files, each with 10 reads.
# Quality control and filtering {#sec-qc-filtering}This chapter explains how to use [fmicompbio/SingleMoleculeGenomicsIO]{.githubpkg} to filter single molecule footprinting data.The package contains functionality to calculate a collection of quality statistics for each read, and use these to filter out low-quality reads. This filtering can be done either on the imported `SummarizedExperiment` object (both for `modBam` and `mismatchBam` file types), or directly on the bam file (currently only `modBam`, which will generate another, filtered `modBam` file).[fmicompbio/SingleMoleculeGenomicsIO]{.githubpkg} also provides several ways to filter out individual genomic positions based on, e.g. the genomic sequence or the read coverage. We start by loading the required packages. In addition to the software package, we load a `BSgenome` object providing the mouse genome sequence.```{r}#| message: false#| label: load-packagesBSgenomeName <-"BSgenome.Mmusculus.GENCODE.GRCm39.gencodeM34"library(SingleMoleculeGenomicsIO)library(BSgenomeName, character.only =TRUE)library(SummarizedExperiment)library(SparseArray)# Load genomegnm <-get(BSgenomeName)genome(gnm) <-"mm39"```<!-- ## Creating a QC report -->## Position filteringTo illustrate the position-level filtering of imported data, we first read 6mA data from a small genomic region for two samples.We add the sequence context (a single nucleotide) to be able to use this information as a basis for filtering.```{r}#| label: read-datase <-readModBam(bamfiles =c(wt1 ="data/mESC_wt_6mA_rep1.bam",wt2 ="data/mESC_wt_6mA_rep2.bam"),modbase ="a", regions ="chr8:39286301-39287100", seqinfo =seqinfo(gnm), sequenceContextWidth =1, sequenceReference = gnm)se```The sequence information is stored in the `rowData` of `se`: ```{r}#| label: show-rowdatarowData(se)```Position filtering can now be performed with the [filterPositions]{.fnio} function. The `filters` argument define which filters to apply, as well as the order (sequence context, coverage, removal of positions without non-NA values).Here, we retain only positions where the genome sequence is an A, and the coverage (the number of overlapping reads) is at least five. The `assayNameCov` argument indicates which assay will be used to define the coverage. If this is a read-level assay (like here), coverage will first be calculated using [flattenReadLevelAssay]{.fnio}. For more precise control, the summary assay can also be manually calculated and added to `se` beforehand, and specified in `assayNameCov`.```{r}#| label: filter-positionssefilt <-filterPositions( se, filters =c("sequenceContext", "coverage", "all.na"),sequenceContext ="A",assayNameCov ="mod_prob",minCov =5)sefilt```In this case, the position filtering reduced the number of unique positions from `r nrow(se)` to `r nrow(sefilt)` by `r sprintf("%.1f%%", 100 * (nrow(se) - nrow(sefilt)) / nrow(se))`. However, the number of non-NA values in the matrix is only reduced from `r sum(is_nonna(as.matrix(assay(se, "mod_prob"))))` to `r sum(is_nonna(as.matrix(assay(sefilt, "mod_prob"))))` (`r sprintf("%.1f%%", 100 * (sum(is_nonna(as.matrix(assay(se, "mod_prob")))) - sum(is_nonna(as.matrix(assay(sefilt, "mod_prob"))))) / sum(is_nonna(as.matrix(assay(se, "mod_prob")))))`), confirming that the filtered-out positions are generally covered only by few reads. In addition to explicit filtering shown above, several functions in [fmicompbio/SingleMoleculeGenomicsIO]{.githubpkg} and [fmicompbio/footprintR]{.githubpkg} allow on-the-fly filtering for a specific sequence context (for example [calcReadStats]{.fnio} or [plotRegion]{.fn}). ## Read filtering### Filtering a `SummarizedExperiment` objectIn addition to the position filtering illustrated above, [fmicompbio/SingleMoleculeGenomicsIO]{.githubpkg} also provides functionality for calculating read-level quality scores and filtering out reads with low quality. The calculation of the quality scores is done using the [addReadStats]{.fnio} function, and the filtering is performed via the [filterReads]{.fnio} function.```{r}#| label: fig-filter-reads# Calculate read statisticssefilt <-addReadStats( sefilt, name ="QC")# The calculated read statistics are stored in the colDatacolData(sefilt)sefilt$QC$wt1# In addition, we can filter based on the read info columns added by readModBamsefilt$readInfo$wt1# Visualize the read statistics to set appropriate filter thresholdsplotReadStats( sefilt)# Perform filteringsefilt <-filterReads( sefilt,minQscore =13,maxFracLowConf =0.1,minAlignedLength =5000, removeAllNApos =TRUE)```Filtering statistics are stored in the `metadata` of the filtered `SummarizedExperiment` object.```{r}#| label: show-metadatametadata(sefilt)$filteredOutReads```### Filtering a `modBam` fileAs mentioned above, [fmicompbio/SingleMoleculeGenomicsIO]{.githubpkg} can also be used to directly filter alignments in a `modBam` file.The [filterReadsModBam]{.fnio} function reads the alignments in the input file, parses them, and writes them to the output file if they pass all the designated filters. These filters are treated hierarchically - in other words, if a read does not pass a given filter, the remaining filters will not be examined and the processing continues with the next read.Here we illustrate the `modBam` filtering using two small example `modBam` files, each with 10 reads. ```{r}#| label: filter-bamfile# input filesmodbamfiles <-c("data/6mA_1_10reads.bam", "data/6mA_2_10reads.bam")# output filesfiltbamfiles <-sub("\\.bam", "_filtered.bam", modbamfiles)res <-filterReadsModBam(infiles = modbamfiles,outfiles = filtbamfiles,modbase ="a",indexOutfiles =FALSE,minReadLength =6746,minAlignedLength =6896,minAlignedFraction =0.56,minQscore =9.7,maxFracLowConf =0.11,maxEntropy =0.29,verbose =TRUE)```The `res` object provides details about the number of reads that were filtered out at each stage. ```{r}#| label: show-filter-bamfileres``````{r}#| echo: false#| label: remove-filtered-bamfilesunlink(filtbamfiles)```## Session info<details><summary><b>Click to view session info</b></summary>```{r}#| label: session-infosessioninfo::session_info(info ="packages")```</details>