Filter out features in PD/TMT data
filterPDTMT.RdIf inputLevel is "Proteins", exclude features with
'Score.Sequest.HT.Sequest.HT' below minScore,
'Number.of.Peptides' below minPeptides, or identified as
'Contaminant' by ProteomeDiscoverer.
If inputLevel is "PeptideGroups", exclude features with
'Delta.Score.by.Search.Engine.Sequest.HT' below minDeltaScore,
'Number.of.PSMs' below minPSMs, or identified as
'Contaminant' by ProteomeDiscoverer.
filterPDTMT(
sce,
inputLevel,
minScore = 0,
minPeptides = 0,
minDeltaScore = 0,
minPSMs = 0,
masterProteinsOnly = FALSE,
modificationsCol = "Modifications",
excludeUnmodifiedPeptides = FALSE,
keepModifications = NULL,
plotUpset = TRUE,
exclFile = NULL
)Arguments
- sce
A
SummarizedExperimentobject (or a derivative).- inputLevel
Either
"Proteins"or"PeptideGroups", indicating the type of features insce.- minScore
Numeric scalar, the minimum allowed value in the 'Score.Sequest.HT.Sequest.HT' column in order to retain the feature. Only used if
inputLevelis"Proteins".- minPeptides
Numeric scalar, the minimum allowed value in the 'Number.of.Peptides' column in order to retain the feature. Only used if
inputLevelis"Proteins".- minDeltaScore
Numeric scalar, the minimum allowed value in the 'Delta.Score.by.Search.Engine.Sequest.HT' column in order to retain the feature. Only used if
inputLevelis"PeptideGroups".- minPSMs
Numeric scalar, the minimum allowed value in the 'Number.of.PSMs' column in order to retain the feature. Only used if
inputLevelis"PeptideGroups".- masterProteinsOnly
Logical scalar indicating whether only master proteins (where the
Mastercolumn value isIsMasterProtein) should be retained.- modificationsCol
Character string pointing to a column containing modification details.
excludeUnmodifiedPeptidesandkeepModificationswill use information from this column. Only used ifinputLevelis"PeptideGroups".- excludeUnmodifiedPeptides
Logical scalar, whether to filter out peptides without modifications. Only used if
inputLevelis"PeptideGroups".- keepModifications
Character string (or
NULL) indicating which modifications to retain in the analysis. Can be a regular expression, which will be matched against themodificationsCol. IfNULL(the default), all rows are retained. Only used ifinputLevelis"PeptideGroups".- plotUpset
Logical scalar, whether to generate an UpSet plot detailing the reasons for features being filtered out. Only generated if any feature is in fact filtered out.
- exclFile
Character scalar, the path to a text file where the features that are filtered out are written. If
NULL(default), excluded features are not recorded.
Value
A filtered object of the same type as sce.
Examples
## Proteins
sce <- importExperiment(
inFile = system.file("extdata", "pdtmt_example",
"Fig2_m23139_RTS_QC_varMods_Proteins.txt",
package = "einprot"),
iColPattern = "^Abundance.F.+.Sample.")$sce
dim(sce)
#> [1] 1723 16
sce <- filterPDTMT(sce = sce, inputLevel = "Proteins", minScore = 2,
minPeptides = 2, plotUpset = TRUE)
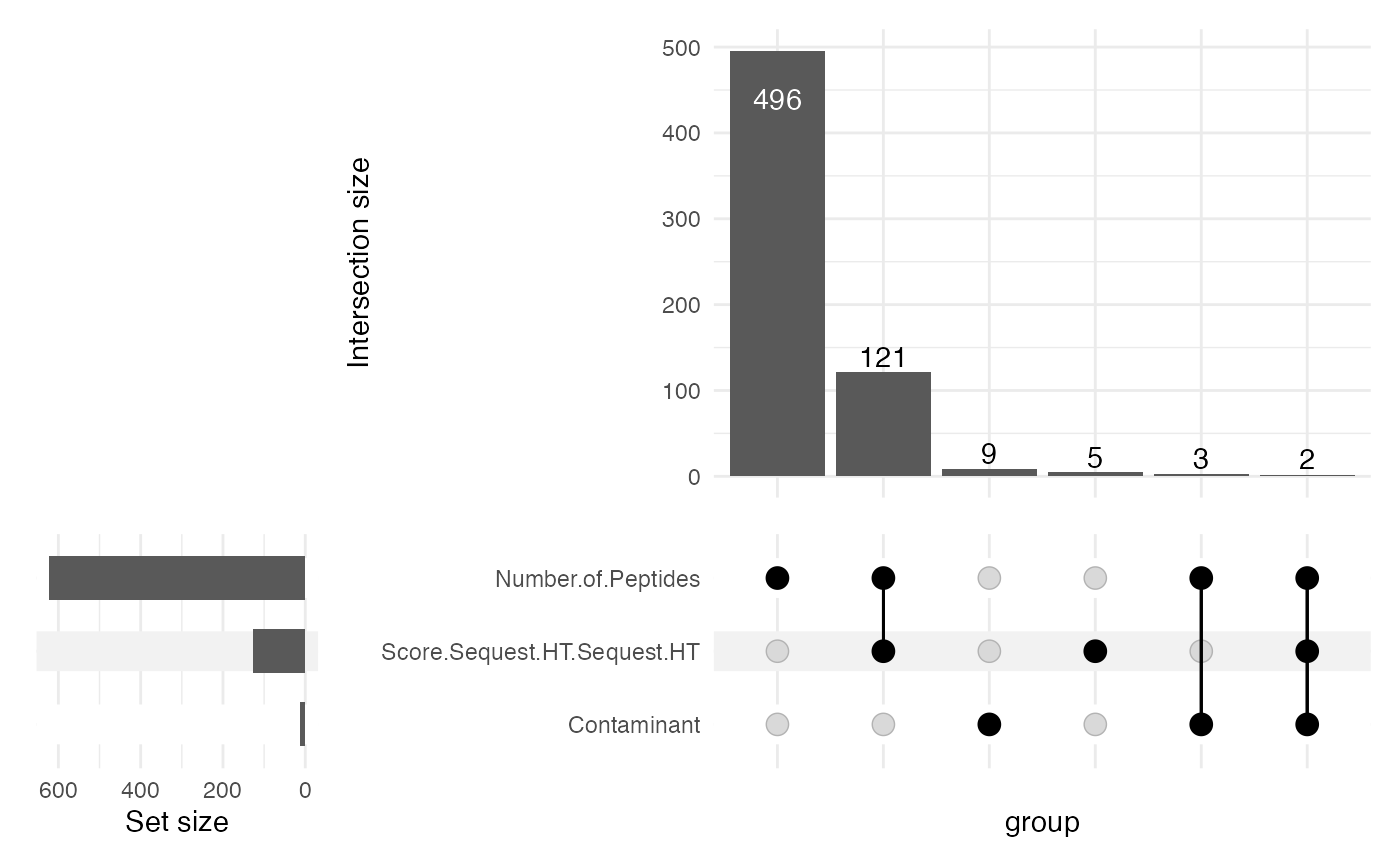 dim(sce)
#> [1] 1087 16
## PeptideGroups
sce <- importExperiment(
inFile = system.file("extdata", "pdtmt_example",
"Fig2_m23139_RTS_QC_varMods_PeptideGroups.txt",
package = "einprot"),
iColPattern = "^Abundance.F.+.Sample.")$sce
dim(sce)
#> [1] 5756 16
sce <- filterPDTMT(sce = sce, inputLevel = "PeptideGroups",
minPSMs = 2, plotUpset = TRUE, minDeltaScore = 0.2,
modificationsCol = "Modifications.in.Master.Proteins",
excludeUnmodifiedPeptides = TRUE)
dim(sce)
#> [1] 1087 16
## PeptideGroups
sce <- importExperiment(
inFile = system.file("extdata", "pdtmt_example",
"Fig2_m23139_RTS_QC_varMods_PeptideGroups.txt",
package = "einprot"),
iColPattern = "^Abundance.F.+.Sample.")$sce
dim(sce)
#> [1] 5756 16
sce <- filterPDTMT(sce = sce, inputLevel = "PeptideGroups",
minPSMs = 2, plotUpset = TRUE, minDeltaScore = 0.2,
modificationsCol = "Modifications.in.Master.Proteins",
excludeUnmodifiedPeptides = TRUE)
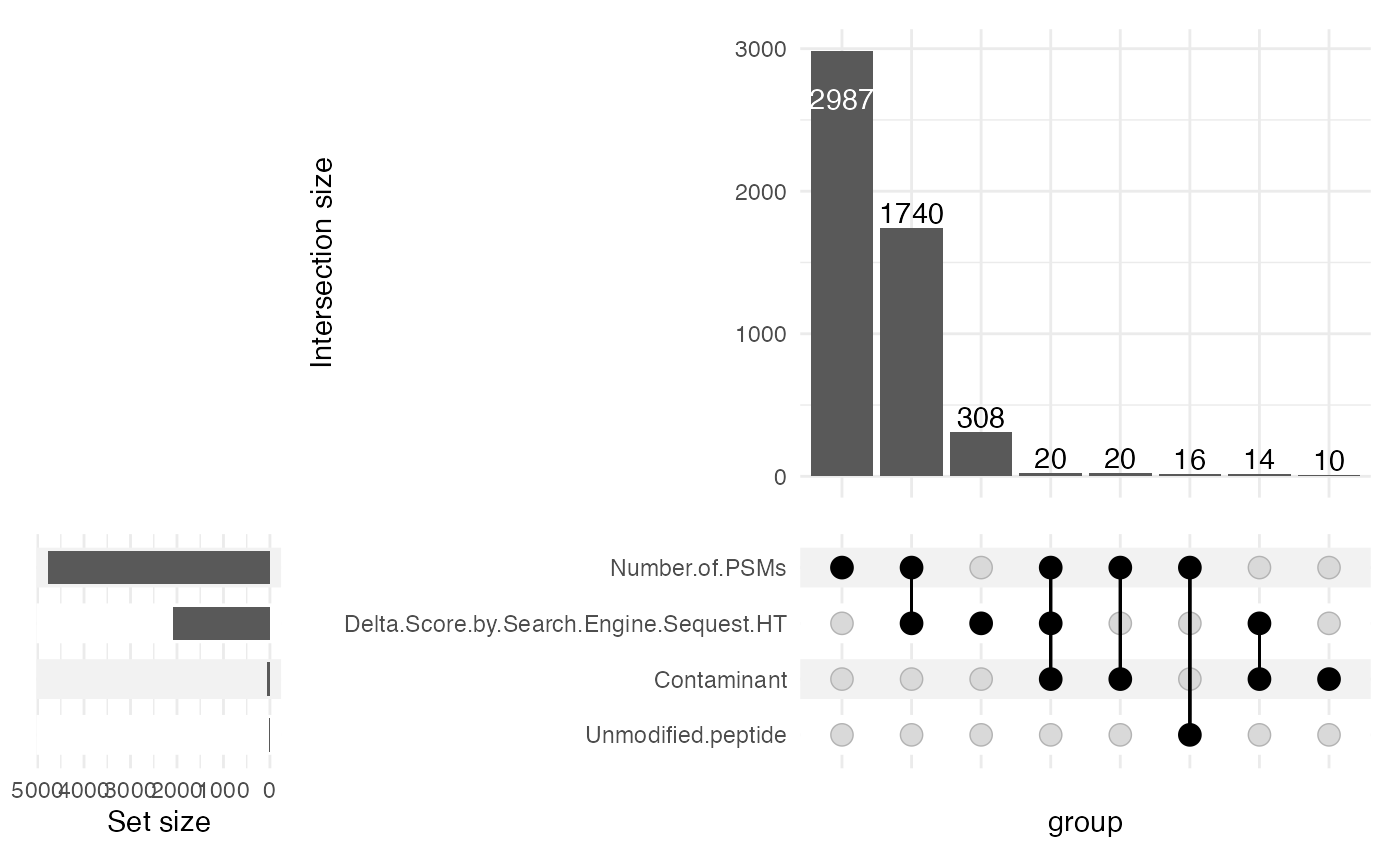 dim(sce)
#> [1] 641 16
dim(sce)
#> [1] 641 16