TabulaMurisSenisData
Charlotte Soneson, Dania Machlab, Federico Marini
28 April 2026
Source:vignettes/TabulaMurisSenisData.Rmd
TabulaMurisSenisData.Rmd
library(SingleCellExperiment)
library(TabulaMurisSenisData)
library(ggplot2)Introduction
This package provides access to the processed bulk and single-cell
RNA-seq data from the Tabula Muris Senis data set (Schaum et al. 2019; Tabula Muris Consortium
2020). The processed bulk RNA-seq data was downloaded from GEO
(accession number GSE132040).
The single-cell data (droplet and FACS) was obtained from FigShare (see
below for links). All data sets are provided as
SingleCellExperiment objects for easy access and use within
the Bioconductor ecosystem.
Bulk RNA-seq data
The bulk RNA-seq data can be accessed via the
TabulaMurisSenisBulk() function. By setting the argument
infoOnly to TRUE, we can get information about
the size of the data set before downloading it.
tmp <- TabulaMurisSenisBulk(infoOnly = TRUE)
#> Total download size: 59.8 MiB
tms_bulk <- TabulaMurisSenisBulk()
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
tms_bulk
#> class: SingleCellExperiment
#> dim: 54352 947
#> metadata(0):
#> assays(1): counts
#> rownames(54352): 0610005C13Rik 0610006L08Rik ... n-TSaga9 n-TStga1
#> rowData names(8): source type ... havana_gene tag
#> colnames(947): A1_384Bulk_Plate1_S1 A1_384Bulk_Plate3_S1 ...
#> P9_384Bulk_Plate2_S369 P9_384Bulk_Plate3_S369
#> colData names(19): Sample name title ... __alignment_not_unique organ
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):We list the available tissues.
Single-cell RNA-seq data
Droplet data
The data files for the droplet single-cell RNA-seq data were downloaded from FigShare:
We list the available tissues.
listTabulaMurisSenisTissues(dataset = "Droplet")
#> [1] "All" "Large_Intestine" "Pancreas" "Trachea"
#> [5] "Skin" "Fat" "Thymus" "Liver"
#> [9] "Heart_and_Aorta" "Mammary_Gland" "Bladder" "Lung"
#> [13] "Kidney" "Limb_Muscle" "Spleen" "Tongue"
#> [17] "Marrow"As for the bulk data, we can print the size of the data set before downloading it.
tmp <- TabulaMurisSenisDroplet(tissues = "All", infoOnly = TRUE)
#> Total download size (All): 709.0 MiB
tms_droplet <- TabulaMurisSenisDroplet(tissues = "All")
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> require("rhdf5")
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
tms_droplet
#> $All
#> class: SingleCellExperiment
#> dim: 20138 245389
#> metadata(0):
#> assays(1): counts
#> rownames(20138): Xkr4 Rp1 ... Sly Erdr1
#> rowData names(6): n_cells means ... highly_variable varm
#> colnames(245389): AAACCTGCAGGGTACA-1-0-0-0 AAACCTGCAGTAAGCG-1-0-0-0 ...
#> 10X_P8_15_TTTGTCATCGGCTTGG-1 10X_P8_15_TTTGTCATCTTGTTTG-1
#> colData names(15): age cell ... louvain leiden
#> reducedDimNames(2): PCA UMAP
#> mainExpName: NULL
#> altExpNames(0):We plot the UMAP of the entire data set and color by tissue, to re-create the plot from here.
# tissue colors
tissue_cols <- c(Pancreas = "#3182bd", Thymus = "#31a354",
Trachea = "#636363", Bladder = "#637939",
Lung = "#7b4173", Large_Intestine = "#843c39",
Fat = "#969696", Tongue = "#a1d99b",
Mammary_Gland = "#ce6dbd", Limb_Muscle = "#d6616b",
Marrow = "#de9ed6", Skin = "#e6550d",
Liver = "#e7969c", Heart_and_Aorta = "#e7ba52",
Kidney = "#e7cb94", Spleen = "#fd8d3c")
# get dataset with all tissues
se <- tms_droplet$All
se
#> class: SingleCellExperiment
#> dim: 20138 245389
#> metadata(0):
#> assays(1): counts
#> rownames(20138): Xkr4 Rp1 ... Sly Erdr1
#> rowData names(6): n_cells means ... highly_variable varm
#> colnames(245389): AAACCTGCAGGGTACA-1-0-0-0 AAACCTGCAGTAAGCG-1-0-0-0 ...
#> 10X_P8_15_TTTGTCATCGGCTTGG-1 10X_P8_15_TTTGTCATCTTGTTTG-1
#> colData names(15): age cell ... louvain leiden
#> reducedDimNames(2): PCA UMAP
#> mainExpName: NULL
#> altExpNames(0):
# prepare data set for ggplot
ds <- as.data.frame(reducedDim(se, "UMAP"))
ds <- cbind(ds, tissue = colData(se)$tissue)
head(ds)
#> UMAP1 UMAP2 tissue
#> AAACCTGCAGGGTACA-1-0-0-0 5.5556602 -10.160711 Tongue
#> AAACCTGCAGTAAGCG-1-0-0-0 2.9584570 -14.145093 Tongue
#> AAACCTGTCATTATCC-1-0-0-0 3.1235533 -14.481063 Tongue
#> AAACGGGGTACAGTGG-1-0-0-0 1.5939721 -14.062417 Tongue
#> AAACGGGGTCTTCTCG-1-0-0-0 0.5233619 -8.997872 Tongue
#> AAAGATGAGCTATGCT-1-0-0-0 1.0210617 -14.642970 Tongue
# plot
ggplot(ds, aes(x = UMAP1, y = UMAP2, color = tissue)) +
geom_point(size = 0.05) +
scale_color_manual(values = tissue_cols) +
theme_classic() +
guides(colour = guide_legend(override.aes = list(size = 5)))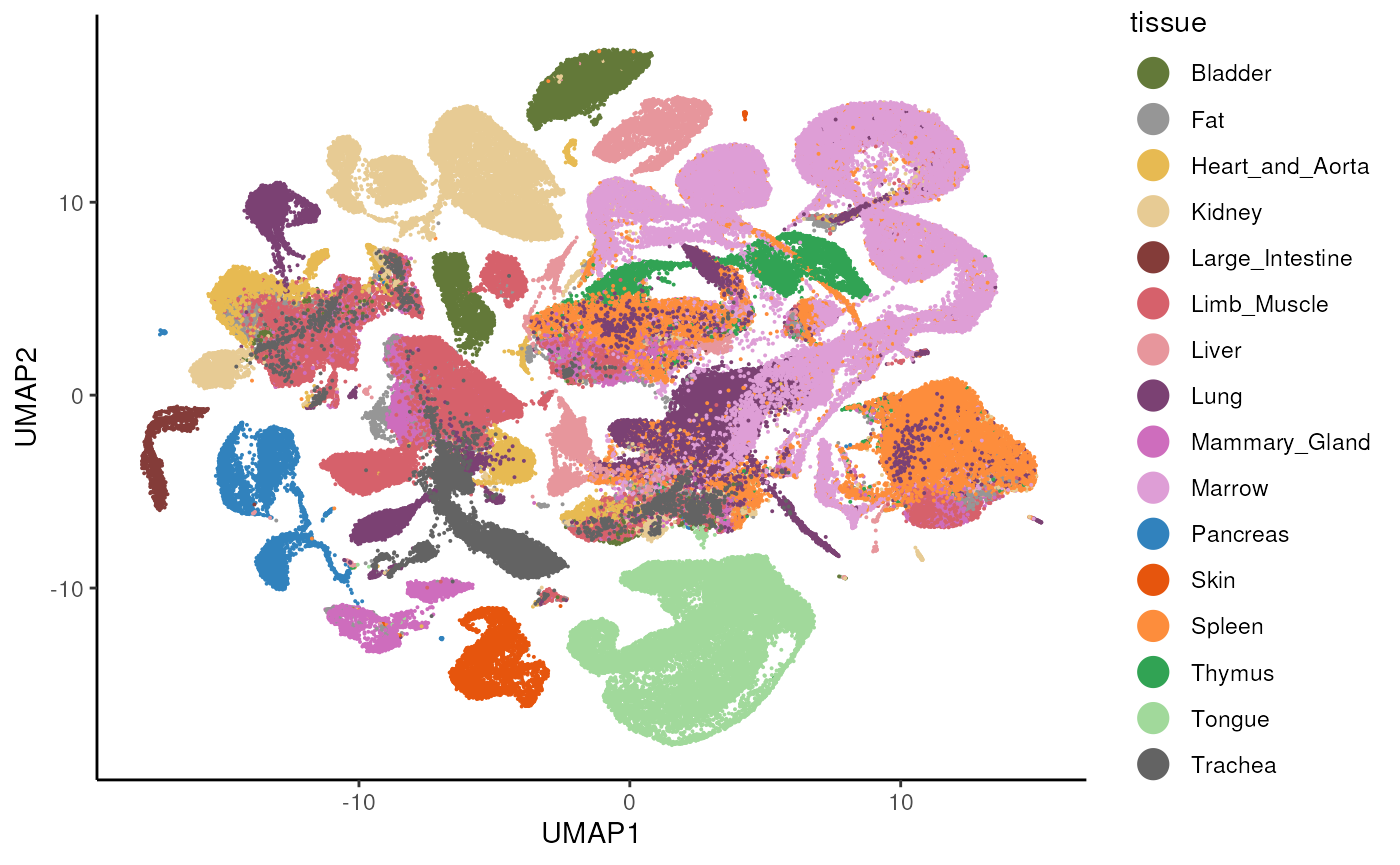
FACS data
The data files for the FACS single-cell RNA-seq data were downloaded from FigShare:
We list the available tissues.
listTabulaMurisSenisTissues(dataset = "FACS")
#> [1] "All" "Aorta" "Kidney"
#> [4] "Diaphragm" "BAT" "Spleen"
#> [7] "Limb_Muscle" "Liver" "MAT"
#> [10] "Thymus" "Trachea" "GAT"
#> [13] "SCAT" "Bladder" "Lung"
#> [16] "Mammary_Gland" "Pancreas" "Skin"
#> [19] "Tongue" "Brain_Non-Myeloid" "Heart"
#> [22] "Brain_Myeloid" "Large_Intestine" "Marrow"Also here, we can print the size of the data set before downloading it.
tmp <- TabulaMurisSenisFACS(tissues = "All", infoOnly = TRUE)
#> Total download size (All): 697.0 MiB
tms_facs <- TabulaMurisSenisFACS(tissues = "All")
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
tms_facs
#> $All
#> class: SingleCellExperiment
#> dim: 22966 110824
#> metadata(0):
#> assays(1): counts
#> rownames(22966): 0610005C13Rik 0610007C21Rik ... Zzef1 Zzz3
#> rowData names(6): n_cells means ... highly_variable varm
#> colnames(110824): A10_B000497_B009023_S10.mm10-plus-0-0
#> A10_B000756_B007446_S10.mm10-plus-0-0 ... P9_B000492_S153.mus-2-1
#> P9_MAA001700_S105.mus-2-1
#> colData names(15): FACS.selection age ... louvain leiden
#> reducedDimNames(2): PCA UMAP
#> mainExpName: NULL
#> altExpNames(0):We plot the UMAP of the entire data set and color by tissue, to re-create the plot from here.
# tissue colors
tissue_cols <- c(Skin = "#e6550d", Pancreas = "#3182bd",
Limb_Muscle = "#d6616b", Heart = "#e7ba52",
Spleen = "#fd8d3c", Diaphragm = "#8c6d31",
Trachea = "#636363", Tongue = "#a1d99b",
Thymus = "#31a354", `Brain_Non-Myeloid` = "#cedb9c",
Brain_Myeloid = "#b5cf6b", Bladder = "#637939",
Large_Intestine = "#843c39", BAT = "#9c9ede",
GAT = "#bd9e39", MAT = "#a55194", SCAT = "#6baed6",
Lung = "#7b4173", Liver = "#e7969c",
Marrow = "#de9ed6", Kidney = "#e7cb94",
Aorta = "#393b79", Mammary_Gland = "#ce6dbd")
# get dataset with all tissues
se <- tms_facs$All
se
#> class: SingleCellExperiment
#> dim: 22966 110824
#> metadata(0):
#> assays(1): counts
#> rownames(22966): 0610005C13Rik 0610007C21Rik ... Zzef1 Zzz3
#> rowData names(6): n_cells means ... highly_variable varm
#> colnames(110824): A10_B000497_B009023_S10.mm10-plus-0-0
#> A10_B000756_B007446_S10.mm10-plus-0-0 ... P9_B000492_S153.mus-2-1
#> P9_MAA001700_S105.mus-2-1
#> colData names(15): FACS.selection age ... louvain leiden
#> reducedDimNames(2): PCA UMAP
#> mainExpName: NULL
#> altExpNames(0):
# prepare data set for ggplot
ds <- as.data.frame(reducedDim(se, "UMAP"))
ds <- cbind(ds, tissue = colData(se)$tissue)
head(ds)
#> UMAP1 UMAP2 tissue
#> A10_B000497_B009023_S10.mm10-plus-0-0 9.917321 10.959189 Skin
#> A10_B000756_B007446_S10.mm10-plus-0-0 12.558255 -14.239025 Pancreas
#> A10_B000802_B009022_S10.mm10-plus-0-0 10.272095 9.065876 Skin
#> A10_B000927_B007456_S10.mm10-plus-0-0 1.349453 7.980347 Limb_Muscle
#> A10_B001361_B007505_S10.mm10-plus-0-0 4.771871 -7.167833 Heart
#> A10_B002452_B009020_S10.mm10-plus-0-0 3.218485 -8.522328 Spleen
# plot
ggplot(ds, aes(x = UMAP1, y = UMAP2, color = tissue)) +
geom_point(size = 0.05) +
scale_color_manual(values = tissue_cols) +
theme_classic() +
guides(colour = guide_legend(override.aes = list(size = 5)))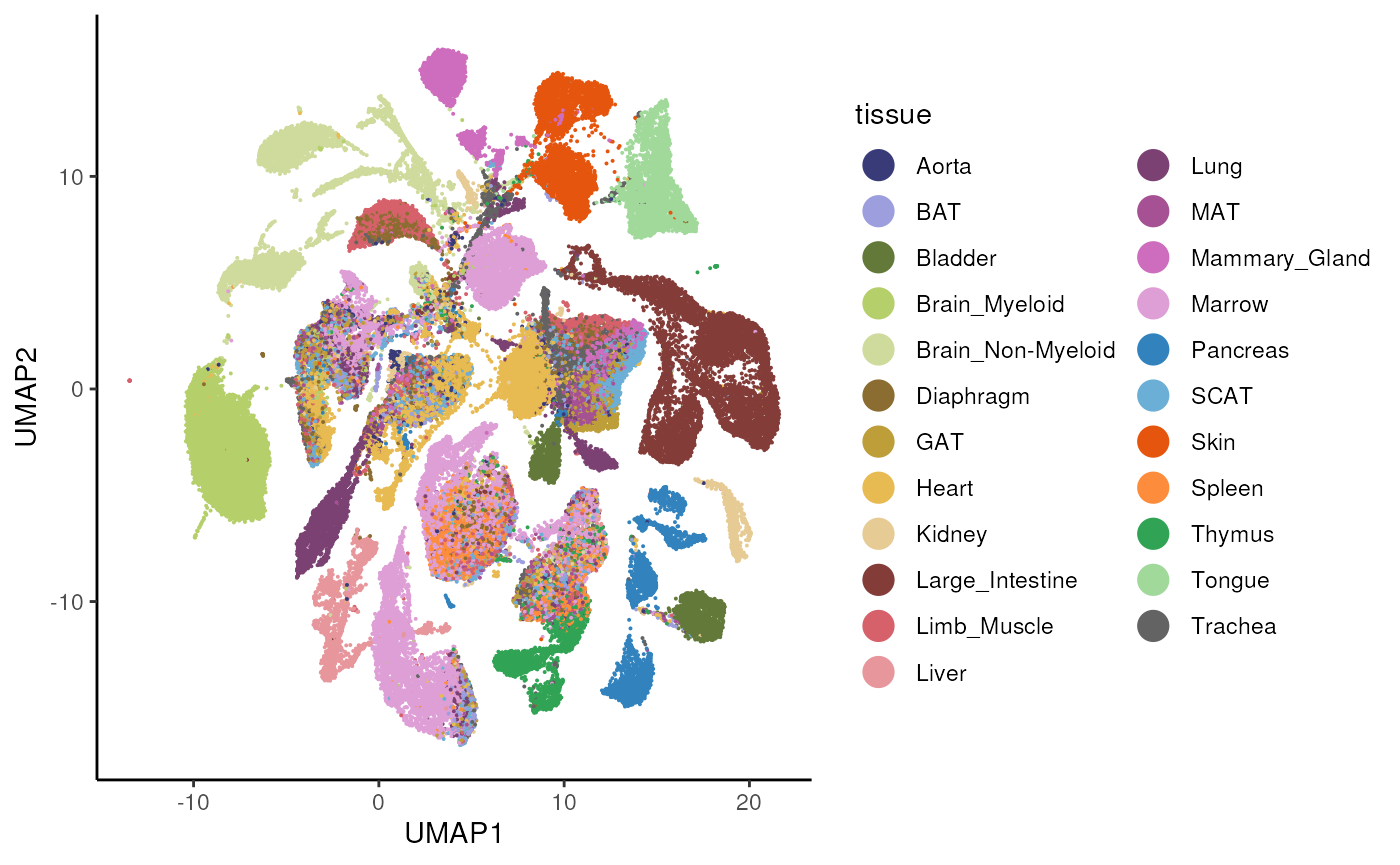
Explore data with iSEE
The Tabula Muris Senis datasets are provided in the form of
SingleCellExperiment objects.
A natural companion to this data structure is the iSEE
package, which can be used for interactive and reproducible data
exploration.
Any analysis steps should be performed in advance before calling
iSEE, and since these datasets can be quite big, the
operations can be time consuming, and/or require a considerable amount
of resources.
sce_all_facs <- TabulaMurisSenisFACS(tissues = "All", processedCounts = TRUE)$All
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
#> see ?TabulaMurisSenisData and browseVignettes('TabulaMurisSenisData') for documentation
#> loading from cache
sce_all_facs
#> class: SingleCellExperiment
#> dim: 22966 110824
#> metadata(0):
#> assays(2): counts logcounts
#> rownames(22966): 0610005C13Rik 0610007C21Rik ... Zzef1 Zzz3
#> rowData names(6): n_cells means ... highly_variable varm
#> colnames(110824): A10_B000497_B009023_S10.mm10-plus-0-0
#> A10_B000756_B007446_S10.mm10-plus-0-0 ... P9_B000492_S153.mus-2-1
#> P9_MAA001700_S105.mus-2-1
#> colData names(15): FACS.selection age ... louvain leiden
#> reducedDimNames(2): PCA UMAP
#> mainExpName: NULL
#> altExpNames(0):We can see that the sce_all_facs contains both raw and
processed counts (in the assays slots counts and
logcounts), but also the PCA and UMAP embeddings as
provided in the original publication.
assayNames(sce_all_facs)
#> [1] "counts" "logcounts"
reducedDimNames(sce_all_facs)
#> [1] "PCA" "UMAP"If desired, additional processing steps can be performed on the
sce_all_facs - e.g., storing signature scores (computed via
AUCell) as
colData elements.
Once these steps are completed, launching iSEE is as easy
as
This will launch iSEE with a standard default set of
panels.
Optionally, we can configure the initial set to start the
app in the desired configuration - below, we show how to start
iSEE with:
- a
ReducedDimensionPlotdisplaying the UMAP, colored by the expression of a feature; - a
RowDataTable, for selecting a feature to be transmitted to the other panels (showing all genes whose name contains “Col1”); - a
FeatureAssayPlot, for showing the expression of the gene selected in theRowDataTable, split by tissue.
initial <- list()
################################################################################
# (Compact) Settings for Reduced dimension plot 1
################################################################################
initial[["ReducedDimensionPlot1"]] <- new(
"ReducedDimensionPlot",
DataBoxOpen = TRUE,
Type = "UMAP",
VisualBoxOpen = TRUE,
ColorBy = "Feature name",
ColorByFeatureName = "Col1a1",
ColorByFeatureSource = "RowDataTable1",
ColorByFeatureDynamicSource = FALSE
)
################################################################################
# (Compact) Settings for Row data table 1
################################################################################
initial[["RowDataTable1"]] <- new(
"RowDataTable",
Selected = "Col1a1",
Search = "Col1"
)
################################################################################
# (Compact) Settings for Feature assay plot 1
################################################################################
initial[["FeatureAssayPlot1"]] <- new(
"FeatureAssayPlot",
DataBoxOpen = TRUE,
Assay = "logcounts",
XAxis = "Column data",
XAxisColumnData = "tissue",
YAxisFeatureName = "Col1a1",
YAxisFeatureSource = "RowDataTable1"
)
Click here to display the complete configuration chunk
initial <- list()
################################################################################
# Settings for Reduced dimension plot 1
################################################################################
initial[["ReducedDimensionPlot1"]] <- new("ReducedDimensionPlot", Type = "UMAP", XAxis = 1L, YAxis = 2L,
FacetRowByColData = "FACS.selection", FacetColumnByColData = "FACS.selection",
ColorByColumnData = "FACS.selection", ColorByFeatureNameAssay = "logcounts",
ColorBySampleNameColor = "#FF0000", ShapeByColumnData = "FACS.selection",
SizeByColumnData = "n_genes", FacetRowBy = "None", FacetColumnBy = "None",
ColorBy = "Feature name", ColorByDefaultColor = "#000000",
ColorByFeatureName = "Col1a1", ColorByFeatureSource = "RowDataTable1",
ColorByFeatureDynamicSource = FALSE, ColorBySampleName = "A10_B000497_B009023_S10.mm10-plus-0-0",
ColorBySampleSource = "---", ColorBySampleDynamicSource = FALSE,
ShapeBy = "None", SizeBy = "None", SelectionAlpha = 0.1,
ZoomData = numeric(0), BrushData = list(), VisualBoxOpen = TRUE,
VisualChoices = "Color", ContourAdd = FALSE, ContourColor = "#0000FF",
PointSize = 1, PointAlpha = 1, Downsample = FALSE, DownsampleResolution = 200,
CustomLabels = FALSE, CustomLabelsText = "A10_B000497_B009023_S10.mm10-plus-0-0",
FontSize = 1, LegendPointSize = 1, LegendPosition = "Bottom",
HoverInfo = TRUE, LabelCenters = FALSE, LabelCentersBy = "FACS.selection",
LabelCentersColor = "#000000", VersionInfo = list(iSEE = structure(list(
c(2L, 5L, 1L)), class = c("package_version", "numeric_version"
))), PanelId = c(ReducedDimensionPlot = 1L), PanelHeight = 500L,
PanelWidth = 4L, SelectionBoxOpen = FALSE, RowSelectionSource = "---",
ColumnSelectionSource = "---", DataBoxOpen = FALSE, RowSelectionDynamicSource = FALSE,
ColumnSelectionDynamicSource = FALSE, RowSelectionRestrict = FALSE,
ColumnSelectionRestrict = FALSE, SelectionHistory = list())
################################################################################
# Settings for Row data table 1
################################################################################
initial[["RowDataTable1"]] <- new("RowDataTable", Selected = "Col1a1", Search = "Col1", SearchColumns = c("",
"", "", "", ""), HiddenColumns = character(0), VersionInfo = list(
iSEE = structure(list(c(2L, 5L, 1L)), class = c("package_version",
"numeric_version"))), PanelId = c(RowDataTable = 1L), PanelHeight = 500L,
PanelWidth = 4L, SelectionBoxOpen = FALSE, RowSelectionSource = "---",
ColumnSelectionSource = "---", DataBoxOpen = FALSE, RowSelectionDynamicSource = FALSE,
ColumnSelectionDynamicSource = FALSE, RowSelectionRestrict = FALSE,
ColumnSelectionRestrict = FALSE, SelectionHistory = list())
################################################################################
# Settings for Feature assay plot 1
################################################################################
initial[["FeatureAssayPlot1"]] <- new("FeatureAssayPlot", Assay = "logcounts", XAxis = "Column data",
XAxisColumnData = "tissue", XAxisFeatureName = "0610005C13Rik",
XAxisFeatureSource = "---", XAxisFeatureDynamicSource = FALSE,
YAxisFeatureName = "Col1a1", YAxisFeatureSource = "RowDataTable1",
YAxisFeatureDynamicSource = FALSE, FacetRowByColData = "FACS.selection",
FacetColumnByColData = "FACS.selection", ColorByColumnData = "age",
ColorByFeatureNameAssay = "logcounts", ColorBySampleNameColor = "#FF0000",
ShapeByColumnData = "FACS.selection", SizeByColumnData = "n_genes",
FacetRowBy = "None", FacetColumnBy = "None", ColorBy = "None",
ColorByDefaultColor = "#000000", ColorByFeatureName = "0610005C13Rik",
ColorByFeatureSource = "---", ColorByFeatureDynamicSource = FALSE,
ColorBySampleName = "A10_B000497_B009023_S10.mm10-plus-0-0",
ColorBySampleSource = "---", ColorBySampleDynamicSource = FALSE,
ShapeBy = "None", SizeBy = "None", SelectionAlpha = 0.1,
ZoomData = numeric(0), BrushData = list(), VisualBoxOpen = FALSE,
VisualChoices = "Color", ContourAdd = FALSE, ContourColor = "#0000FF",
PointSize = 1, PointAlpha = 1, Downsample = FALSE, DownsampleResolution = 200,
CustomLabels = FALSE, CustomLabelsText = "A10_B000497_B009023_S10.mm10-plus-0-0",
FontSize = 1, LegendPointSize = 1, LegendPosition = "Bottom",
HoverInfo = TRUE, LabelCenters = FALSE, LabelCentersBy = "FACS.selection",
LabelCentersColor = "#000000", VersionInfo = list(iSEE = structure(list(
c(2L, 5L, 1L)), class = c("package_version", "numeric_version"
))), PanelId = c(FeatureAssayPlot = 1L), PanelHeight = 500L,
PanelWidth = 4L, SelectionBoxOpen = FALSE, RowSelectionSource = "---",
ColumnSelectionSource = "---", DataBoxOpen = FALSE, RowSelectionDynamicSource = FALSE,
ColumnSelectionDynamicSource = FALSE, RowSelectionRestrict = FALSE,
ColumnSelectionRestrict = FALSE, SelectionHistory = list())
initialiSEE can then be launched with the following
command:
iSEE(sce_all_facs, initial = initial)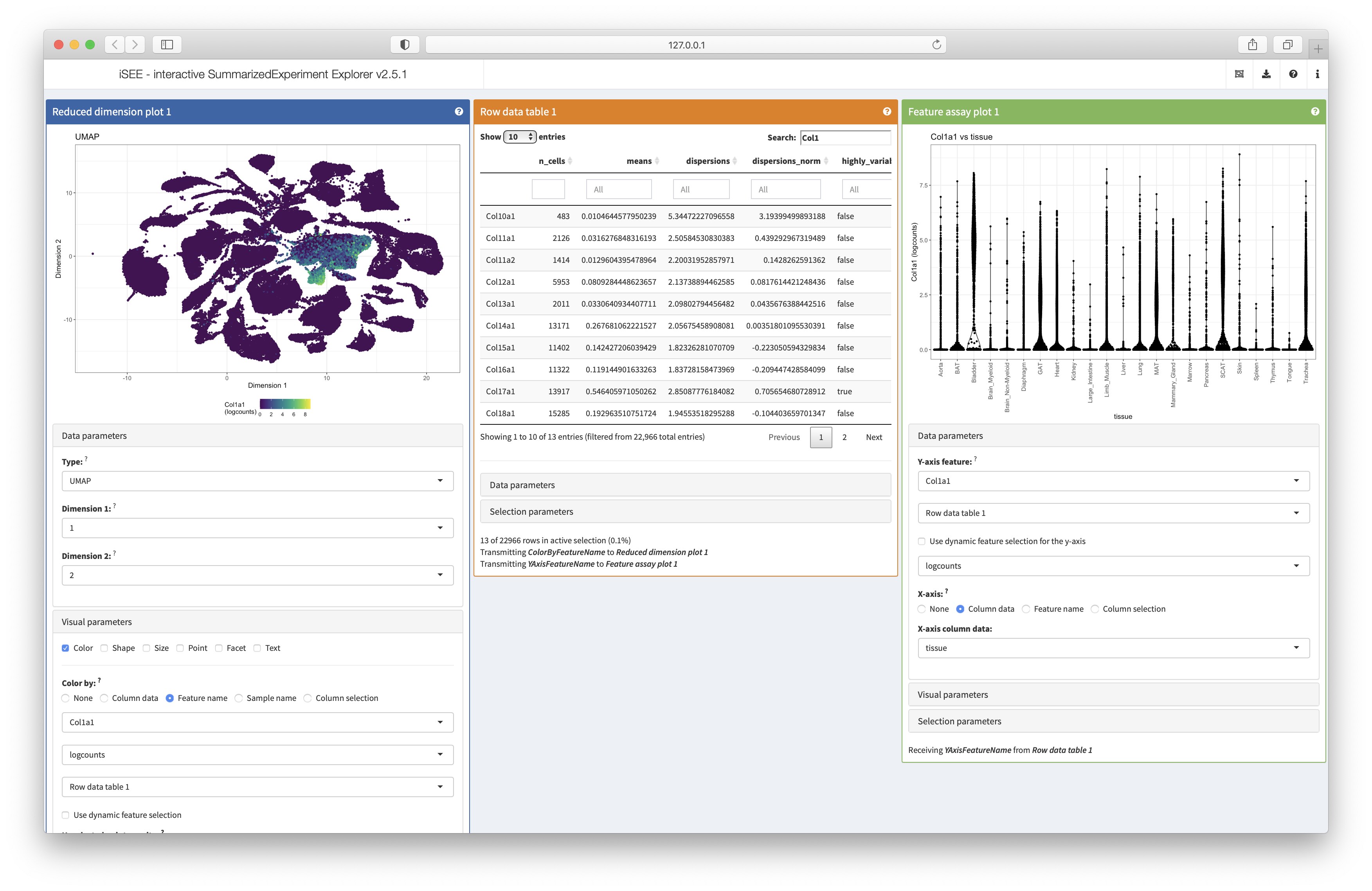
Screenshot of the iSEE app running on the FACS single cell dataset
Note that these lengthy configuration options can be readily exported
by clicking on the dedicated button “Display panel settings” in
iSEE - we can do that anytime after we are done designing
the configuration using the app interface.
Session info
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] iSEE_2.23.1 rhdf5_2.55.16
#> [3] ggplot2_4.0.3 TabulaMurisSenisData_1.19.0
#> [5] SingleCellExperiment_1.33.2 SummarizedExperiment_1.41.1
#> [7] Biobase_2.71.0 GenomicRanges_1.63.2
#> [9] Seqinfo_1.1.0 IRanges_2.45.0
#> [11] S4Vectors_0.49.3 BiocGenerics_0.57.1
#> [13] generics_0.1.4 MatrixGenerics_1.23.0
#> [15] matrixStats_1.5.0 BiocStyle_2.39.0
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 jsonlite_2.0.0 shape_1.4.6.1
#> [4] magrittr_2.0.5 farver_2.1.2 rmarkdown_2.31
#> [7] GlobalOptions_0.1.4 fs_2.1.0 ragg_1.5.2
#> [10] vctrs_0.7.3 memoise_2.0.1 htmltools_0.5.9
#> [13] S4Arrays_1.11.1 AnnotationHub_4.1.0 curl_7.1.0
#> [16] Rhdf5lib_1.99.6 SparseArray_1.11.13 sass_0.4.10
#> [19] bslib_0.10.0 htmlwidgets_1.6.4 desc_1.4.3
#> [22] listviewer_4.0.0 httr2_1.2.2 cachem_1.1.0
#> [25] igraph_2.3.0 mime_0.13 lifecycle_1.0.5
#> [28] iterators_1.0.14 pkgconfig_2.0.3 colourpicker_1.3.0
#> [31] Matrix_1.7-5 R6_2.6.1 fastmap_1.2.0
#> [34] shiny_1.13.0 clue_0.3-68 digest_0.6.39
#> [37] colorspace_2.1-2 AnnotationDbi_1.73.1 ExperimentHub_3.1.0
#> [40] textshaping_1.0.5 RSQLite_2.4.6 filelock_1.0.3
#> [43] labeling_0.4.3 gdata_3.0.1 httr_1.4.8
#> [46] abind_1.4-8 mgcv_1.9-4 compiler_4.6.0
#> [49] bit64_4.8.0 withr_3.0.2 doParallel_1.0.17
#> [52] S7_0.2.2 DBI_1.3.0 shinyAce_0.4.4
#> [55] HDF5Array_1.39.1 rappdirs_0.3.4 DelayedArray_0.37.1
#> [58] rjson_0.2.23 gtools_3.9.5 tools_4.6.0
#> [61] vipor_0.4.7 otel_0.2.0 httpuv_1.6.17
#> [64] glue_1.8.1 h5mread_1.3.3 nlme_3.1-169
#> [67] rhdf5filters_1.23.3 promises_1.5.0 grid_4.6.0
#> [70] cluster_2.1.8.2 gtable_0.3.6 XVector_0.51.0
#> [73] ggrepel_0.9.8 BiocVersion_3.23.1 foreach_1.5.2
#> [76] pillar_1.11.1 later_1.4.8 rintrojs_0.3.4
#> [79] circlize_0.4.18 splines_4.6.0 dplyr_1.2.1
#> [82] BiocFileCache_3.1.0 lattice_0.22-9 bit_4.6.0
#> [85] tidyselect_1.2.1 ComplexHeatmap_2.27.1 Biostrings_2.79.5
#> [88] miniUI_0.1.2 knitr_1.51 bookdown_0.46
#> [91] shinydashboard_0.7.3 xfun_0.57 DT_0.34.0
#> [94] yaml_2.3.12 shinyWidgets_0.9.1 evaluate_1.0.5
#> [97] codetools_0.2-20 tibble_3.3.1 BiocManager_1.30.27
#> [100] cli_3.6.6 xtable_1.8-8 systemfonts_1.3.2
#> [103] jquerylib_0.1.4 Rcpp_1.1.1-1.1 dbplyr_2.5.2
#> [106] png_0.1-9 parallel_4.6.0 pkgdown_2.2.0.9000
#> [109] blob_1.3.0 viridisLite_0.4.3 scales_1.4.0
#> [112] purrr_1.2.2 crayon_1.5.3 GetoptLong_1.1.1
#> [115] rlang_1.2.0 KEGGREST_1.51.1 shinyjs_2.1.1