Calculate the frequencies of same strand alignment distances,
for example from MNase-seq data to estimate nucleosome repeat length.
Distance calculations are implemented in C++ (calcAndCountDist)
for efficiency.
calcPhasogram(fname, regions = NULL, rmdup = TRUE, dmax = 3000L)Arguments
- fname
charactervector with one or several bam files. If multiple files are given, distance counts from all will be summed.- regions
GRangesobject. Only alignments falling into these regions will be used. IfNULL(the default), all alignments are used.- rmdup
logical(1)indicating if duplicates should be removed. IfTRUE(the default), only one of several alignments starting at the same coordinate is used.- dmax
numeric(1)specifying the maximal distance between same strand alignments to count.
Value
integer vector with dmax elements, with the element at
position d giving the observed number of alignment pairs at that
distance.
References
Phasograms were originally described in Valouev et al., Nature 2011 (doi:10.1038/nature10002). The implementation here differs in two ways from the original algorithms:
It does not implement removing of positions that have been seen less than
ntimes (referred to as an-pile subset in the paper).It does allow to retain only alignments that fall into selected genomic intervals (
regionsargument).
See also
estimateNRL to estimate the nucleosome repeat length
from a phasogram, plotPhasogram to visualize an annotated
phasogram, calcAndCountDist for low-level distance counting.
Examples
if (requireNamespace("GenomicAlignments", quietly = TRUE) &&
requireNamespace("Rsamtools", quietly = TRUE)) {
bamf <- system.file("extdata", "phasograms", "mnase_mm10.bam",
package = "swissknife")
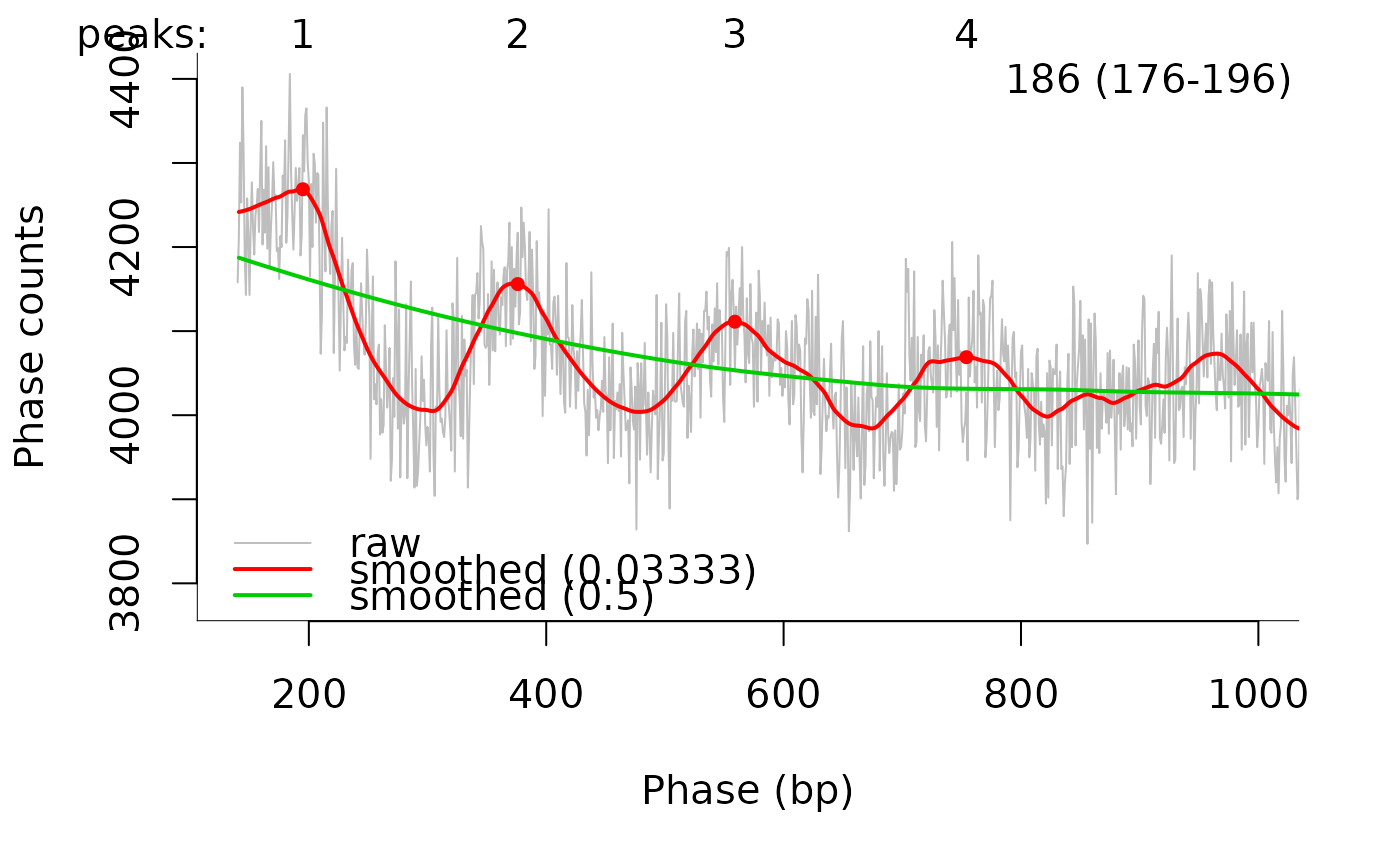
pg <- calcPhasogram(bamf)
print(estimateNRL(pg, usePeaks = 1:4)[1:2])
plotPhasogram(pg, usePeaks = 1:4, xlim = c(0,1000))
}
#> $nrl
#> [1] 186
#>
#> $nrl.CI95
#> 2.5 % 97.5 %
#> 176.0015 195.9985
#>